
配体尺度对配合物结构及光催化降解活性的调节:从零维到一维的Cu(Ⅱ)配合物
2020-03-18 10:35王其宝王海英卢文欣
无机化学学报 2020年2期
王其宝 李 响 王海英 卢文欣*, 王 鹏*,
(1山东科技大学化学与环境工程学院,青岛 266590)
(2四川师范大学化学与材料科学学院,成都 610068)
伴随着配位化学和固态有序晶态材料的性质研究,化学家们合成了大量的功能性金属-有机框架材料(metal-organic frameworks,MOFs),配位化学的研究也从最初的配位小分子领域的磁性、小分子气体吸附、均相催化等性质研究扩展到了配位聚合物尤其是三维MOFs材料领域的二阶非线性光学、可调谐发光、离子交换与识别、选择性分离乃至非均相催化等性质的研究[1-3]。作为一类多孔的复合结晶材料,MOFs近年来在各个研究领域引起了人们的广泛关注,其优异的物理、化学性质(超高比表面积和孔隙率、结构可调节性与修饰性)使其在生物、医药、催化、分离以及荧光检测等许多重要领域中都发挥着重要的作用,而这些性质大多取决于其结构中的孔穴大小及配位的维度,例如三维多孔的MOFs材料往往具有更优良的热稳定性[4]。由于MOFs结构调控的核心是调控配体与金属离子之间的配位作用,所以MOFs的结构调控[5-7]与传统的配位聚合物的结构调控有着相似的影响因素。众所周知,配位聚合物或者MOFs的结构与其性质之间存在关联,因此探讨配位结构的影响因素即使在今天也仍然对合成具有特定性质的MOFs化合物有着非常重要的意义。
随着晶体结构分析技术的日趋成熟,配合物的结构影响因素的研究随之而来。通过前人大量的研究,目前公认的MOFs的结构调节主要是通过配体结构、温度、溶剂、金属种类、金属配体比例等因素的改变而实现,而对于同种金属中心的配合物而言,配合物结构主要受到温度,溶剂和配体结构的影响[8-11]。金属离子与配体的配位原子之间通过配位作用相互连接形成配位键,而配体连接相邻金属离子时,配体的尺度会影响配位聚合物乃至MOFs的结构[12-18]。研究表明,配体的尺度不仅能调节所得MOFs的孔穴大小(如Yaghi等研究组报道的一系列MOFs 化合物[8,10,19]),还可以在尺度发生过大变化的情况下改变配合物的结构,并在第二配体的协助下实现结构的变化(如 Zhou,Mohamed 等[20-22]的工作)。
结构的改变会导致化合物性质发生相应的改变,比如零维的配位大环化合物与一维线型、二维平面或者三维结构的配位聚合物相比,在溶解度、催化反应的效率、对客体分子的包结等方面存在显著的不同,往往具有选择性包结或特殊的催化活性等[23-24]。考虑到上述方面的不同,在我们前期报道的2,6-二(3′-吡啶乙炔基)-4-甲基苯胺(L1)合成工作的基础上[25],我们进一步使用Suzuki偶联反应合成了尺度更短但配位构型相似的桥联吡啶类配体2,6-二(3′-吡啶基)-4-甲基苯胺(L2)。L1和L2两个配体均为桥联吡啶结构,不同的是对甲苯胺与吡啶环之间是否存在其他原子:L1是对甲苯胺通过2个乙炔结构而L2则是使用单键直接连接了2个吡啶,其配位方式、配位角度等较为相似。2个配体尺度的不同会导致弯折型联吡啶配体的伸展方向、空间阻力等均有较大差别,相对于直线型的联吡啶或对苯二甲酸等配体具有更多的调节结构的可能。考虑到阴离子的调控作用会导致L1能与不同的Co(Ⅱ)金属盐配位获得不同维度的配位聚合物[26],我们思考固定金属盐的阴离子种类以考察配体尺度对配合物的结构调节作用,合成基本结构相似但配位维度不同的配位聚合物,试图通过分析所得产物的结构和光催化反应的活性来分析配合物的结构、性质与配体尺度之间的关联情况。在此,我们将报道这2个尺度不同的配体与醋酸铜(Cu(OAc)2·H2O)所形成的不同维度的配合物:“2+2”结构的配位大环分子[Cu2(L1)(OAc)4]2(1)和一维的配位聚合物{[Cu2(L2)(OAc)4]·2CH2Cl2·CH3CN}n(2)。通过常温下的溶剂扩散法合成并获得其单晶后,使用X射线单晶衍射技术获得了这2个化合物的单晶结构,进一步使用红外及元素分析证明了其中的结构信息,探讨了配体尺度对配位聚合物的结构影响。2个配合物与对应配体的固态荧光测试结果证明了铜金属离子强烈的荧光猝灭作用,这导致其与亚甲基蓝(MB)溶液混合后不影响MB分子自身的荧光发射和紫外吸收。鉴于铜配合物可以对MB分子的光催化降解过程有强烈的促进作用[27-29],我们通过MB溶液在可见光条件下的光催化降解实验,对2个化合物作为光降解过程催化剂的活性进行了探索,获得了大环结构比一维聚合物结构具有更强的光催化降解活性的结论,证明配体的尺度对构建的配合物的结构具有重要的影响,并能够影响其光催化降解活性,这对配合物的光催化降解研究具有一定的指导意义。
1 实验部分
1.1 仪器和试剂
二氯甲烷、乙腈、二异丙基胺、乙醇等溶剂均为市售分析纯,采购后蒸馏后使用。吡啶-3-硼酸、3-吡啶乙炔、碘化亚铜、二(三苯基膦)氯化钯、四(三苯基膦)钯、碳酸钾、一水合醋酸铜等金属盐和试剂为商业化试剂,采购后直接使用。2,6-二溴对苯甲胺、配体 L1 参考文献方法合成[25]。
红外光谱使用Nicolet IS50傅立叶变换红外光谱仪。元素分析使用Perkin-Elemental 2400元素分析仪测定。晶体测定使用Bruker Smart APEX CCD单晶衍射仪在常温下收集衍射数据。固体荧光发射性质使用HITACHI F-4600型荧光光谱仪完成。光降解实验用光源为CEL-LPH120型低压汞灯光源系统。紫外-可见吸收使用上海元析UV9000型紫外可见分光光度计测试获得数据。
1.2 配体L2的合成
2,6-二溴对苯甲胺(2.67 g,10 mmol),吡啶-3-硼酸 (3.08 g,25 mmol),四 (三 苯 基 膦 )钯 (1.4 g,1.25 mmol,5%),碳酸钾(7 g,50 mmol),乙醇、1,4-二氧六环各60 mL于250 mL三口瓶中,氮气保护下加热回流50 h,分液,上层红棕色液体旋干,硅胶柱层析(V二氯甲烷∶V四氢呋喃=5∶1) 得浅黄色固体 1.52 g,产率58%,熔点:135~136 ℃。IR(KBr,cm-1):3 387,3 319(NH2),3 015(Ar-H),2 903(CH3),1 644,1 578,1 459,1 421,1 294,1 274,1 251(苯环及吡啶环),810(芳环1,2,4,6 取代)。1H NMR(CDCl3,300 MHz):δ 8.71(s,2H),7.47(d,J=4.3 Hz,2H),7.27(m,J=4.3 Hz,2H),7.19(d,J=4.3 Hz,2H),7.00(s,2H),3.75(s,2H),2.33(s,3H)。元素分析C17H15N3(%,括号内为理论值):C,77.34(78.13);H,5.95(5.79);N,16.91(16.08)。
1.3 配合物1的合成
将配体L1(61.9 mg,0.2 mmol)溶解于二氯甲烷(20 mL)中,将所得淡黄色溶液缓缓加入含有醋酸铜(89.8 mg,0.45 mmol)的 35 mL甲醇溶液中,加热煮沸完全溶解,混合均匀后静置于室温中缓慢挥发至有淡蓝绿色晶体析出时取晶体进行结构测试,剩余部分继续挥发至近干,过滤并用少量二氯甲烷-甲醇混合液洗涤得到1的蓝绿色片状晶体 (73.3 mg,72%)。IR(KBr,cm-1):3 446(m),3 354(m),2 964(w),2 198(m),1 607(s),1 462(s),1 388(m),1 052(m),814(m),614(m)。元素分析C58H54Cu4N6O16(%,括号内为理论值):C,51.34(51.78);H,4.25(4.05);N,6.29(6.25)。
1.4 配合物2的合成
在试管中将含有醋酸铜(41.9 mg,0.21 mmol)的10 mL乙腈溶液铺于溶解配体 L2(26.1 mg,0.1 mmol)的二氯甲烷(10 mL)上,室温下静置至有淡蓝绿色晶体析出,取出测定晶体结构,剩余部分继续反应至结晶完全(约需2周),过滤并用少量二氯甲烷-乙腈混合液洗涤,得到2的蓝绿色片状晶体(59.9 mg,65%)。IR(KBr,cm-1):3 446(m),3 354(m),2 964(w),2 198(m),1 607(s),1 462(s),1 388(m),1 052(m),814(m),614(m)。元素分析 C29H34Cl4Cu2N4O8(%,括号内为理论值):C,41.25(41.69);H,4.33(4.10);N,6.78(6.71)。
1.5 催化光降解实验
分别称量8 mg的晶体1、2与醋酸铜固体,和0.5 mL 30%双氧水溶液各加入100 mL浓度为20 mg·L-1的MB稀溶液中。超声震荡混合均匀后,置于低压汞灯下搅拌,每间隔15 min取样,测其在664 nm处的吸光度。以所测最大紫外吸收无峰时为反应终止时间。以所测吸光度/最初吸光度为纵坐标,以测试时间为横坐标作图进行比较。
1.6 配合物1和2晶体结构的测定
选取透明无裂缝的单晶1(尺寸0.36 mm×0.28 mm×0.08 mm)和 2(尺寸 0.5 mm×0.3 mm×0.3 mm),在298 K(1)或 173 K(2)的温度下,置于 Bruker公司SMART APEX CCD单晶衍射仪上,用经石墨单色化的 Mo Kα(λ=0.071 073 nm)辐射、以 φ-ω 扫描方式收集衍射点,衍射数据使用SAINT软件经Lp因子还原处理并采用SADABS[30]软件进行多重扫描吸收校正后获得 HKL 文件,以 SHELXS软件[31],采用直接法解出配合物晶体结构,并经差值Fourier合成法得到全部非氢原子坐标;用SHELXL程序[32]对非氢原子坐标及其各向异性热参数进行全矩阵最小二乘法精修至收敛;氢原子通过理论加氢方法获得并采用跨式模型(riding modal)进行精修。晶体学数据列于表1中。配合物1和2的主要键长和键角列于表2,配合物2的氢键列于表3。
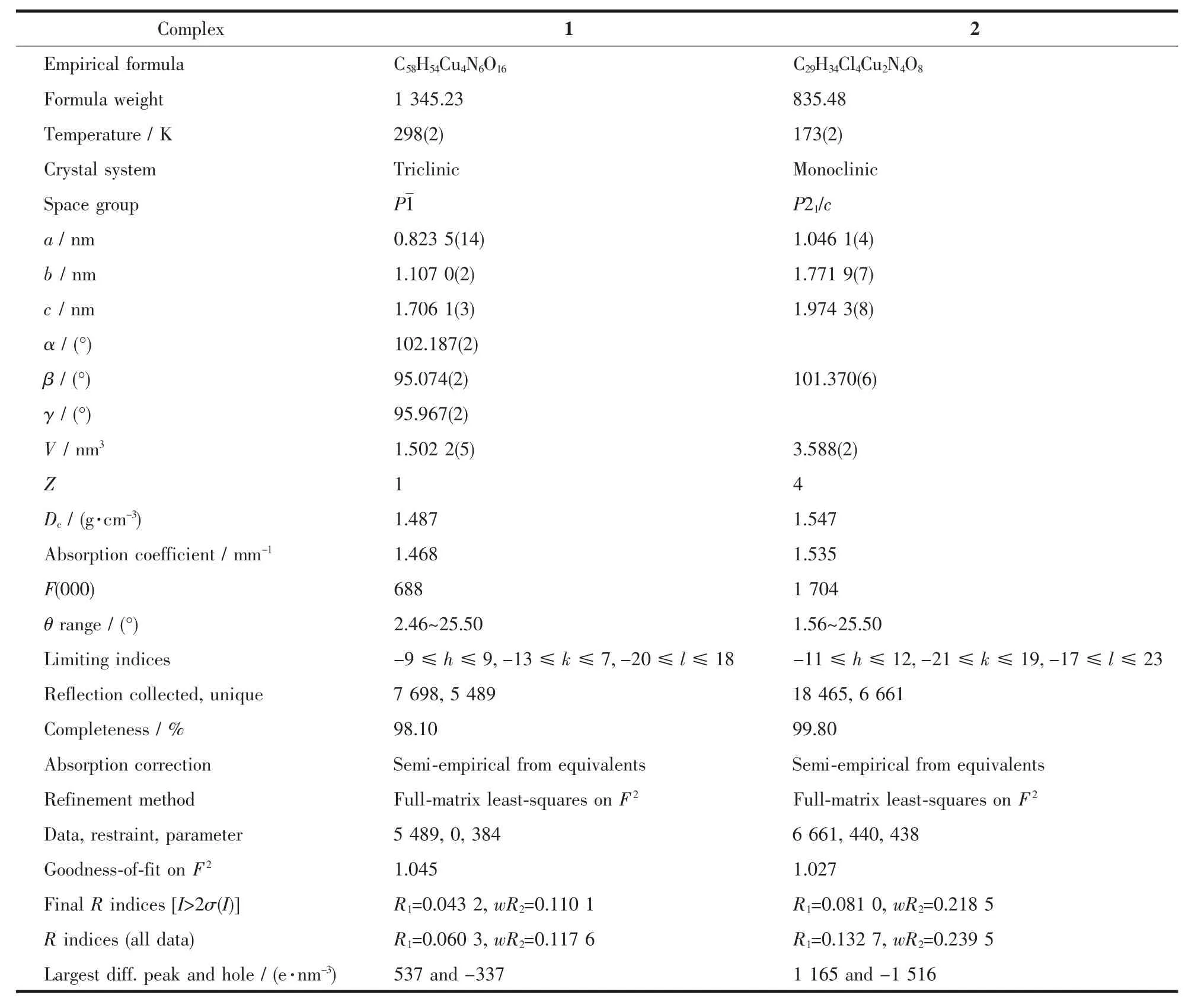
表1 配合物1和2的晶体学数据Table 1 Crystallographic data for complexes 1 and 2

表2 配合物1和2的部分键长(nm)与键角(°)Table 2 Selected bond lengths(nm)and bond angles(°)for complexes 1 and 2

续表2

表3 配位聚合物2的氢键键长和键角Table 3 Hydrogen bond lengths(nm)and bond angles(°)for complex 2
CCDC:1937501,1;1937323,2。
2 结果与讨论
2.1 配合物1的晶体结构
配合物1结晶于三斜晶系P1空间群,晶态下一个不对称结构单元中包含1个配体L1,2个Cu(Ⅱ)离子和4个醋酸根离子,如图1所示。中心Cu(Ⅱ)离子处于一个氮原子,4个氧原子和另一个铜离子的包裹中,位于{CuNO4}的八面体构型的六配位环境的中心。相邻的2个Cu(Ⅱ)离子以二聚物的{Cu2(OAc)4}簇合物形式与2个配体分子键合形成配位大环化合物。配位大环分子上的各个芳香环几乎处于同一个平面内:2个吡啶环之间的平面夹角3.73°,苯环与2个吡啶环之间夹角分别为8.93°(N1所在吡啶环)和6.63°(N2所在吡啶环)。相邻的大环分子之间存在偏移π-π堆积作用:配体苯环所在平面相互平行(二面角为0°)构成 π 平面(由 C8、C9、C10、C12、C13、C14构成,质心坐标 0.812 7,-0.160 5,0.043 5),相邻 2个π平面的面-面距离0.346 nm,质心间距0.501 nm。结合大环分子内部3个几乎处于平面的芳香环,大环分子依靠这些π-π相互作用偏移堆积形成三维结构,如图2所示。
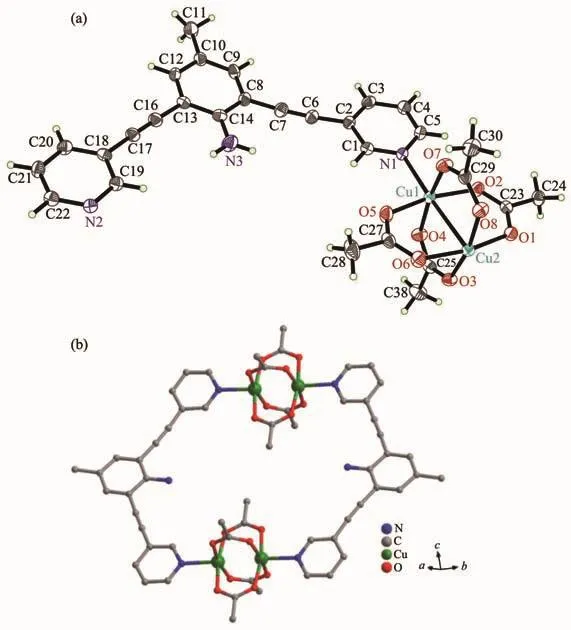
图1 配合物1的椭球度30%的不对称结构单元(a)和大环结构(b)Fig.1 Asymmetric unit with thermal ellipsoid 30%probability level(a)and macrocycle structure(b)of complex 1
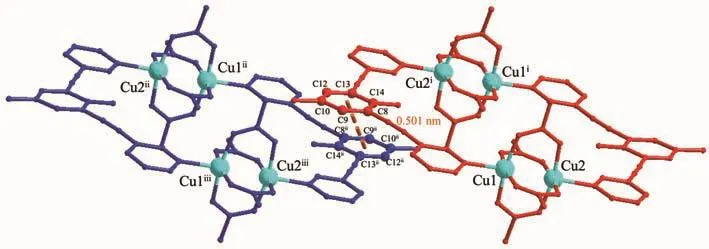
图2 配合物1中相邻配位大环化合物间的π-π堆积作用Fig.2 π-π stacking structures in complex 1
相邻的2个配位大环之间不存在经典的氢键相互作用,仅通过π-π堆积作用相互连接成三维的空间结构,晶体中没有出现大的孔穴,也没有溶剂分子(二氯甲烷或甲醇)的出现。
2.2 配位聚合物2的晶体结构
配位聚合物2结晶于单斜晶系P21/c空间群,晶态下一个不对称结构单元中包含1个配体L2,2个Cu(Ⅱ)离子和4个醋酸酸根离子,晶体中有2个二氯甲烷溶剂分子和1个乙腈溶剂分子,如图3a所示。与1类似,2中的中心Cu(Ⅱ)离子也处于1个氮原子,4个氧原子和1个铜离子的{CuNO4}的八面体构型的六配位环境的中心。相邻的2个Cu(Ⅱ)离子以{Cu2(OAc)4}簇合物的形式存在,并被配体连接形成一维长链结构,如图3b所示。相邻的长链之间存在一组N2-H2A…N4的经典氢键和N2-H2A…Cl1及N2-H2B…Cl2ii的非经典氢键,如图4所示。这些氢键将溶剂分子固定在一维长链上,使得晶体中的溶剂分子较为稳定的存在于长链之间,形成了图5所展示的结构。
对比配位大环化合物1和一维配位聚合物2的结构,由于吡啶环与苯环之间的三键被替换成单键,尺度变小,使得苯环上的氨基对2个吡啶环的阻碍作用变大,导致2个吡啶环不能够再处于同一平面,只能向不同的方向扭曲(2中配体上的2个吡啶环之间的夹角为79.94°,远远大于1中配体上3.73°的夹角)。这种扭曲作用使得配位聚合物2无法形成环状结构而获得一维链状的聚合结构。两者的合成条件相似,仅配体尺度发生改变就导致配位聚合物的维度发生了巨大改变,这说明改变配体的尺度可以对配合物结构造成影响,进而影响其性质,如光催化降解性质以及潜在的催化产生自由基的性质。
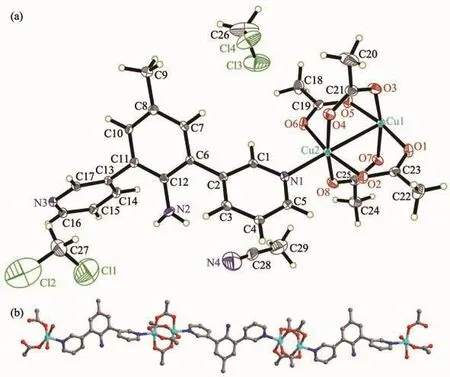
图3 配位聚合物2的椭球度30%的不对称结构单元(a)和忽略氢原子及溶剂分子的一维链状结构(b)Fig.3 Asymmetric unit with thermal ellipsoid 30%probability level(a)and 1D chain structure omitting H atoms and solvent molecules(b)of complex 2
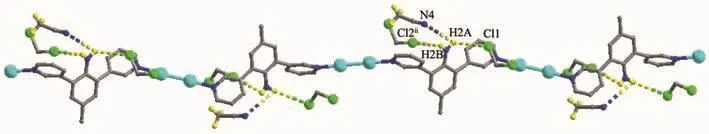
图4 一维长链上以氢键相连的溶剂分子Fig.4 Hydrogen bonds of solvent molecules with 1D chain in compound 2
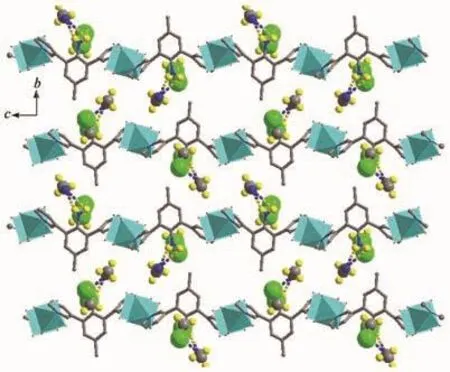
图5 沿晶体学a轴观察到的配位聚合物2中的溶剂分子Fig.5 Solvent molecules in complex 2 viewed along crystallography a axis
2.3 配合物1和2的固态荧光发射性质
我们对所获得的化合物1和2的粉末进行了固态荧光测试,使用350 nm的激发波长获得化合物1和2的固态荧光发射图如图6所示,其结果显示化合物1和2的荧光发射均存在有明显荧光猝灭现象,符合铜配合物常有的荧光猝灭规律[33-34]。
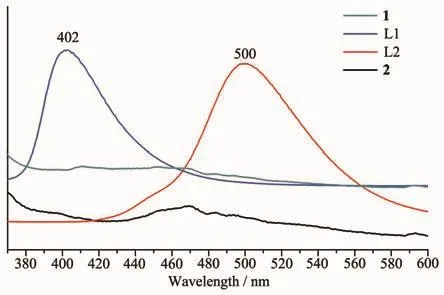
图6 配合物1和2与相应配体的固态荧光发射对比图Fig.6 Luminescence spectra of complexes 1,2 and ligands L1,L2
2.4 配合物1和2对亚甲基蓝的光催化降解性质
根据文献报道,铜基MOFs或者配合物具有光催化降解有机物的性质[35],考虑到不同维度的配合物在溶液中的分散程度不同,我们猜测配合物1和2在溶液中有可能具有不同的光催化活性。基于此,我们采用常见的染料MB为目标,用配合物1和2进行了光催化降解实验,并与纯醋酸铜作为光催化剂的体系进行了对比,以亚甲基蓝的最大紫外吸收作为检测的基准,通过664 nm处的紫外吸光度的变化表征其光催化效果。
如图6所示,在含0.5 mL 30%的双氧水的亚甲基蓝稀溶液中加入8 mg化合物1或2或者醋酸铜的固体后超声震荡,每15 min取样检测1次。其结果显示,配位大环化合物1具有显著的光催化效果,配位聚合物2虽然也展现了优于纯醋酸铜的光催化效果,但是其催化效果远弱于配位大环化合物1,这说明我们合成的配合物均有光催化降解效果,且通过调控配合物的维度能够调节其光催化降解效率。
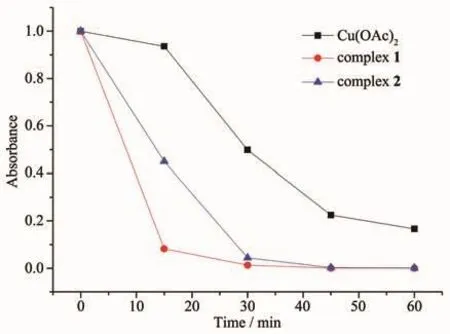
图7 配合物1和2与醋酸铜对亚甲基蓝催化光降解活性对比Fig.7 Comparison of photocatalytic activity of complexes 1 and 2 and Cu(OAc)2for methylene blue
对比实验验证了配位大环化合物在光催化降解方面的活性强于一维配位聚合物的特点,也同时证明催化是由配位的吡啶氮-铜簇配位结构而非醋酸铜结构来实现的。
3 结 论
配合物1和2的结构表明,配体的尺度决定了配合物的结构。配体L1与醋酸铜配位获得的是环状大分子结构,而L2则与醋酸铜形成一维链状结构。由于Cu(Ⅱ)离子存在荧光猝灭的作用,2个铜配合物的固态荧光相比于配体具有明显的猝灭作用。在对配合物1和2的光催化降解性质的探索中,我们发现配位大环化合物1光催化降解亚甲基蓝的活性明显优于一维配位聚合物2,两者均强于醋酸铜的催化光降解性质。
猜你喜欢
科学导报(2022年21期)2022-04-10
新疆医科大学学报(2020年1期)2020-03-12
北京大学学报(自然科学版)(2019年6期)2019-11-27
人工晶体学报(2019年5期)2019-06-18
当代陕西(2019年6期)2019-04-17
分析化学(2018年2期)2018-03-02
成长·读写月刊(2017年3期)2017-04-08
科技视界(2016年26期)2016-12-17
药学研究(2015年11期)2015-12-19
中学科技(2014年11期)2014-12-25
