
MKS1 基因变异所致Joubert 综合征1 例临床及基因分析
2020-03-12 04:04:08张广宇李三松王明梅赵云霞朱登纳
临床儿科杂志 2020年2期
张广宇 李三松 杨 磊 王明梅 赵云霞 朱登纳
郑州大学第三附属医院儿童康复科(河南郑州 450052)
Joubert 综合征于 1969 年首次报道,是一种罕见的颅脑先天发育畸形,其发病率约为1/100000,遗传方式主要为常染色体隐性遗传,少数为X连锁遗传[1-2]。Joubert 综合征的临床表现包括新生儿期的阵发性呼吸过度或呼吸暂停、发育迟缓、眼球运动障碍(包括追视障碍、眼球震颤和斜视等)、肌张力减低、共济失调等,常合并视网膜、肾脏、肝脏等多器官受累[3]。本文回顾分析1例MKS1基因突变所致Joubert 综合征患儿的临床特点和基因检测结果。
1 临床资料
患儿,男,3 个月,因双眼不追视就诊。患儿系G1P1,足月顺产,无宫内窘迫及出生窒息史,出生体质量3.5 kg,Apgar评分不详。生后数天因呼吸急促于当地医院住院治疗20余天,诊断为“肺炎、败血症”。母孕期体健,否认毒物、放射线接触史。父母体健,否认近亲婚配,家系中无类似病史。体格检查:身高60 cm,体质量5.5 kg,头围39.6 cm,前囟 1.5 cm×1.5 cm;神清,反应好,全身皮肤、浅表淋巴结无异常;双侧瞳孔正大等圆,对光反射灵敏,双眼不追随物体移动,无明显眼球震颤、斜视,不追听;心肺腹未见异常;抬头差,四肢肌张力偏低,双膝腱反射引出,病理反射未引出。实验室检查:血常规、肝肾功能、心肌酶谱、甲状腺功能未见异常;氨基酸和酰基肉碱谱分析示丙酰肉碱轻度增高,尿气相色谱质谱有机酸检测未见异常。彩超示肝脏、胆囊、脾脏、肾脏、输尿管、膀胱未见明显异常。眼底检查未见视网膜色素变性,裂隙灯检查未见虹膜缺如。头颅磁共振(MRI)示小脑蚓部发育不良,小脑半球间见“中线裂征”,小脑上脚增粗、变长与中脑呈“磨牙征“,第四脑室呈“蝙蝠翼状”(图1)。临床诊断:Joubert综合征。
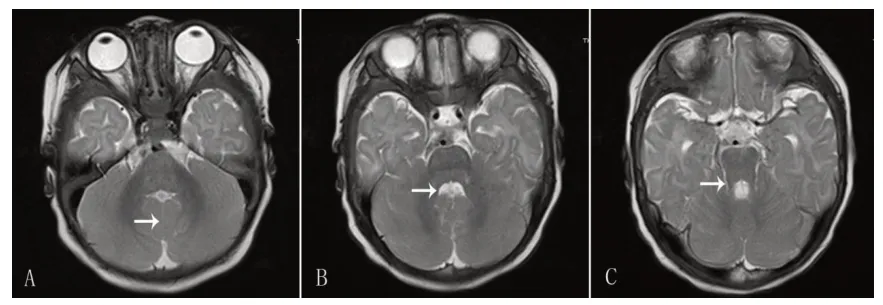
图1 患儿头颅磁共振表现
为进一步明确诊断,经医学伦理审核及家长知情同意,对患儿及父母行全外显子检测和Sanger测序验证。结果发现,17号染色体上的MKS1基因存在c.1411_c.1412 insG 移码突变及c.44 A>G 错义突变。其中,c.1411_c.1412 insG 移码突变是在16 号外显子区的1411-1412位碱基之间插入1个鸟嘌呤,导致其蛋白质翻译在第471 位谷氨酸发生移码变成甘氨酸,并在第591位终止编码(p.E471Gfs*120)。c.1411_c.1412insG位点在dbSNP 数据库中的编号为 rs 762668200,在ExAc的数据库中显示其突变频率为0.0012,ClinVar数据库将其定义为致病性变异。c.44A>G错义突变位于1 号外显子区的第44 位,由腺嘌呤突变为鸟嘌呤,可导致其蛋白质翻译在第15 位酪氨酸变成半胱氨酸(p.Y15C),DYDF、dbSNP、千人基因组、千人南方、千人北方、ExAC中均未见收录。通过生物信息学软件,Provean、Polyphen2、Sift、Mutationtaster、M-CAP,对蛋白功能预测为有害突变;根据ACMG指南,c.44A>G位点的致病性等级为可能致病。Sanger测序验证结果表明,患儿c.1411_c.1412insG移码突变来自于父亲,而c.44 A>G 错义突变来自于母亲(图2),符合常染色体隐性遗传共分离规律。由此判定,MKS 1基因的c.1411_c.1412insG以及c.44A>G的复合杂合突变为患儿的致病性突变,并确诊为Joubert综合征。
入院后给予康复治疗7个疗程。1岁时行Gesell发育量表评估示大运动95分,精细动作90分,适应性90分,语言92分,个人-社交95分。2岁时随访,神清,反应好,追视欠灵活,能说简单句子,能听懂简单指令,能识别五官,认识家人,竖头稳,双手抓物灵活,能指尖捏物,能独站、独走,四肢肌张力偏低。
2 讨论
Joubert 综合征是一种遗传性先天性小脑性共济失调,其特征是中脑-后脑发育不良,MRI表现“磨牙征”和多种器官受累。目前Joubert综合征诊断标准包括:①头颅MRI检查的轴向视图上的“磨牙征”,表现为发育不良的小脑蚓部、加深的脚间窝和增厚且变长的小脑上脚,在周围脑脊液衬托下,轴位上双侧小脑上脚与中脑交接处呈“磨牙”样改变;②不同程度的智力缺陷/发育迟缓;③婴儿期的肌张力减退;④以下1 种或2 种表现(非必要条件但支持诊断),婴儿期不规则呼吸模式(发作性呼吸暂停和/或呼吸急促,有时交替)和眼球运动异常(眼球震颤、斜视、追视障碍等)[1,3-6]。
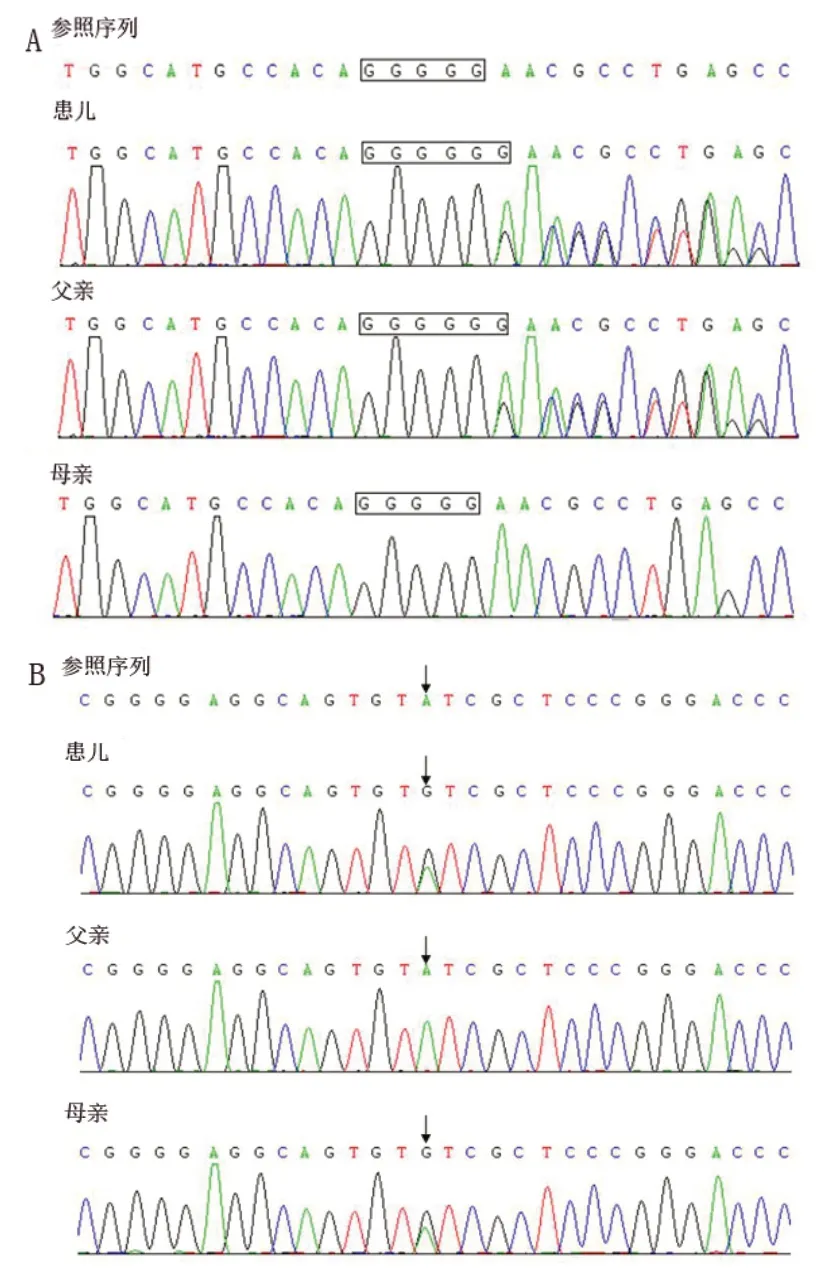
图2 患儿及父母MSK1 基因突变位点Sanger 测序图
Joubert综合征的临床表现可能因受累器官不同而不同,并且脏器受累婴儿期可无相应表现,随患儿年龄增长逐渐显现。在这些缺陷中,最常见的是视网膜缺陷(严重程度从Leber 先天性黑曚到缓慢进展性视网膜病,视力部分保留),肾缺陷(肾单位肾痨或多囊肾)和先天性肝纤维化。较少见的特征包括脉络膜视网膜或视神经缺损,先天性心脏畸形,内脏反转,严重脊柱侧弯,骨骼发育不良,先天性巨结肠症以及口腔和面部缺损,如唇裂、腭裂或两者并存,分叶舌,舌或口腔软组织肿瘤等。由于Joubert综合征临床表现的复杂性,将所有MRI 图像具有“磨牙征”的疾病统称为“Joubert 综合征及相关疾病”,并将其分为单纯型Joubert综合征、Joubert综合征伴眼睛缺陷、Joubert 综合征伴肾脏缺陷、Joubert 综合征伴眼肾缺陷、Joubert综合征伴肝脏缺陷、Joubert综合征伴口面指/趾缺陷[4-5,7]。
本例患儿存在发育迟缓、肌张力降低、眼球运动异常,MRI 有典型的“磨牙征”,无眼睛、肝脏、肾脏受累表现,也无肢体发育畸形,考虑为单纯型 Joubert综合征。全外显子检测发现患儿携带了MKS 1基因c.1411_c.1412insG移码突变和c.44A>G 错义突变,而Sanger测序验证表明移码突变来源于父亲、错义突变来源于母亲,符合常染色体隐性遗传共分离规律。因此判定MKS1基因的c.1411_c.1412insG以及c.44A>G的复合杂合变异为本例Joubert 综合征患儿的致病性变异位点。
Joubert综合征致病与初级纤毛的功能障碍有关,因此Joubert综合征也被纳入初级纤毛疾病之列。初级纤毛是一种不能移动的细胞器,几乎存在于所有细胞类型的表面。虽然长期以来被认为是一个功能的残余物,但这种细胞器最近成为深入研究的焦点,它可能在胚胎发育、人类的遗传性疾病甚至肿瘤发生中发挥关键作用。初级纤毛从基底体出现,是一种固定在细胞质膜上的中心粒结构,其结构核心是轴丝,由9个双管微管组成。在许多成体组织中,初级纤毛起到细胞外信号的传感器作用,并在细胞内转导这个信号以调节组织稳态、增殖等。例如,在肾和胆管上皮或视网膜光感受器中,如果纤毛的传感器功能破坏就会导致相应的器官缺陷。此外研究表明,大多数类型的神经细胞都具有初级纤毛,因此可能在大脑发育和功能中发挥作用[8-12]。
MKS 1基因突变是Meckel 综合征的致病基因之一。Meckel综合征是一种罕见的常染色体隐性遗传性疾病,也是初级纤毛相关疾病的一种,主要临床表现为囊性肾发育不良、严重的中枢神经系统畸形(枕部脑膜脑膨出最常见)、多指和肝脏纤维化改变[13]。近几年报道MKS1基因突变会导致Joubert综合征[14-16]。MKS1基因在组织中广泛表达,尤其在脑、肝,肾中表达量很高。MKS1基因编码的MKS1蛋白含559个氨基酸多肽,其含有高度保守的B9结构域,定位于纤毛基底体[13]。在肾上皮细胞系模型中,通过siRNA瞬时敲除MKS1基因,抑制中心粒向顶膜的迁移,从而导致初级纤毛形成障碍,说明MKS1基因是初级纤毛形成所必需的[17]。MKS1基因突变既会导致Meckel综合征也会导致Joubert综合征,同一基因突变可以出现不同疾病表型可能与基因突变类型有关,如Meckel综合征基因突变类型多为2 个无义突变,而Joubert 综合征至少有1个不会导致蛋白截短的突变(如错义突变)[15]。Meckel综合征以及Joubert综合征均属于初级纤毛相关疾病,它们的基因、表型互相重叠、交织,目前发现Joubert 综合征有30 余个致病基因,其中有10 余个基因的突变会导致Meckel综合征[18-19]。
目前对于Jouber综合征尚无特效治疗,主要为对症治疗及康复训练。Joubert 综合征预后不一,某些基因类型、合并器官受累以及合并其他畸形会导致患者死亡风险增加[20]。Jouber综合征多存在中-重度智力障碍,仅部分患儿智力轻度障碍甚至可能完全正常[4]。本例患儿康复治疗后智力运动发育商达到正常同龄儿童水平,考虑与其突变基因类型以及早期康复介入有关。
综上,本例Joubert 综合征患儿经全外显子组测序及Sanger 测序验证,检测到MKS 1基因的2 个突变c.1411_c.1412insG和c.44A>G,其中c.44A>G为新发现变异,这一发现拓宽了 Joubert 综合征致病基因的突变谱。
猜你喜欢
广东药科大学学报(2023年5期)2023-12-30 00:08:39
实用医院临床杂志(2023年4期)2023-07-31 01:57:48
自然杂志(2022年3期)2022-08-18 03:00:06
时代报告·奔流(2022年1期)2022-04-29 04:10:56
昆明医科大学学报(2022年3期)2022-04-19 13:59:42
中国临床解剖学杂志(2021年2期)2021-04-19 14:52:46
创新作文(小学版)(2019年4期)2019-07-24 09:03:42
中华皮肤科杂志(2019年5期)2019-06-24 06:32:06
哲思2.0(2017年12期)2017-03-13 17:45:04
西南军医(2016年2期)2016-01-23 02:14:10
