
AVPR2 基因变异致先天性肾性尿崩症2 例报告并文献复习
2020-03-12 04:04:04王传凯王春林
临床儿科杂志 2020年2期
王传凯 王春林 梁 黎
浙江大学医学院附属第一医院(浙江杭州 310000)
先天性肾性尿崩症(congenial nephrogenic diabetes insipidus,CNDI)是一组遗传异质性疾病,其发病机制为精氨酸加压素受体及受体后信号转导障碍,使肾远曲小管对精氨酸加压素不敏感导致尿液浓缩功能障碍,临床以多饮多尿及尿比重低为特点。目前报道的致病基因主要为精氨酸加压素受体2(arginine vasopressin receptor 2,AVPR2)基因或水通道蛋白2基因(aquaporin-2,AQP-2)基因,其中AVPR2基因突变所致的CNDI多为X连锁隐性遗传,约占所有CNDI的90%以上。本文回顾分析浙江大学医学院附属第一院儿科收治的2例经基因诊断明确的CNDI患儿的临床资料,并复习相关文献。
1 临床资料
例1,男,5岁8个月,因发现多饮、多尿3年余,生长迟缓半年就诊。就诊前3年出现多饮多尿,夜尿2、3次,尿色清如水,无尿路刺激症状,无视物模糊,无反复头痛,无食欲增加,就诊前半年出现生长迟缓。门诊查血钠159 mmol/L,尿比重1.005,遂拟“尿崩症”收入院。患儿系足月顺产,2岁时高热惊厥1次。否认家族遗传病史。体格检查:体温37.2 ℃,心率102次/min,血压106/74 mmHg,身高107.4 cm(P3),体质量19 kg,神清,精神可,无明显脱水貌,两肺呼吸音清,未闻及干湿啰音,心律齐,心音中,未闻及杂音,腹软,肝脾肋下未及,肾区无叩痛,神经系统阴性。实验室检查:血钠149 mmol/L,血钾4.16 mmol/L,尿素氮4.8 mmol/L,血糖7.5 mmol/L,血渗透压318.6 mmol/L,尿比重波动于1.000~1.002 之间,糖化血红蛋白A1c 5.9%。双肾、输尿管B超未见明显异常。住院期间患儿每日水入量约4 016~4 876 mL,每日尿量约3 120~3 700 mL。外院曾行生长激素激发试验(禁食禁水8小时),测血钠159 mmol/L,血钾4.1 mmol/L,尿素氮 4 mmol/L,血糖6.5 mmol/L,血渗透压336 mmol/L,尿比重1.005。排除习惯性多饮,尿崩症诊断明确。入院行直接加压素试验:试验前血钠149 mmol/L,血钾4.2 mmol/L,尿素氮 4.8 mmol/L,血糖7.5 mmol/L,血渗透压319.7 mmol/L;垂体后叶加压素3.8 U 皮下注射,留取3 次小便,测尿比重分别为1.002、1.000、1.000,复测血钠158 mmol/L,血钾4.7 mmol/L,尿素氮 4.7 mmol/L,血糖7.5 mmol/L,血渗透压337.6 mmol/L;因患儿持续低比重尿,血渗透压明显升高,试验停止。给予氢氯噻嗪片及吲哚美辛片口服。1 年后电话随访,患儿体质量23 kg,身高118 cm,身高增长10.6 cm;夜尿明显减少,但总尿量及饮水量仍多,尿比重 1.000,肌酐41 μmol/L,尿素 5.7 mmol/L,钾4.24 mmol/L,钠 142 mmol/L,氯100 mmol/L,提示无肾功能异常及电解质紊乱。随访期间无明显胃肠道症状、无造血系统受抑制等吲哚美辛不良反应。
例2,男,3 岁2 个月,因发现多饮、多尿2 年余就诊。就诊前2年出现多饮多尿,日饮水约3 000 mL,白天尿5、6次,夜尿3次左右,尿色清如水,无反复头痛、呕吐,无视物模糊,无长期低热、盗汗,无食欲增加,无尿频尿急尿痛。拟“尿崩症”收入院。既往史及个人史无特殊。否认家族遗传病史。体格检查:体温36.8 ℃,心率108次/min,血压88/47 mmHg,身高91 cm(P3),体质量14 kg,神清,精神可,无明显脱水貌,两肺呼吸音清,未闻及干湿啰音,心律齐,心音中,未闻及杂音,腹平软,肝脾肋下未及,无肾区叩痛,神经系统阴性。实验室检查:血钠142 mmol/L,血钾4.05 mmol/L,尿素氮3.3 mmol/L,肌酐 35 μmol/L,血糖4.6 mmol/L,血渗透压300 mmol/L,尿比重波动于1.001~1.002之间。双肾、输尿管B超未见明显异常。住院期间患儿每日水入量约2 382 mL,每日尿量约2 848 mL。入院后行禁水-加压素试验:试验前血钠142 mmol/L,血钾4.05 mmol/L,尿素氮 3 mmol/L,血糖4.6 mmol/L,血渗透压299.7 mmol/L;禁食禁饮8小时后,监测尿比重无明显变化(波动于1.001~1.002),血钠159 mmol/L,血钾4.17 mmol/L,尿素氮 3.3 mmol/L,血糖5.9 mmol/L,血渗透压308 mmol/L;予垂体后叶加压素2.6 U皮下注射,留取3 次小便,测尿比重均为1.002,血钠159 mmol/L,血钾4.17 mmol/L,尿素氮 3.3 mmol/L,血糖5.9 mmol/L,血渗透压335.5 mmol/L;因患儿烦躁明显停止试验。给予氢氯噻嗪联合吲哚美辛口服。1年后电话随访:患儿身高增长9.7 cm,尿量及饮水量明显减少,复查尿比重 1.005,肌酐30 μmol/L,尿素5.3 mmol/L,钾3.9 mmol/L,钠143 mmol/L,提示无肾功能异常及电解质紊乱。
经医院医学伦理委员会审批并获取家属知情同意,留取2 例患儿及例2 患儿父母的外周血标本,EDTA 抗凝。DNA 提取、PCR 产物电泳、纯化、测序及序列分析由北京德易东方转化医学研究中心进行。基因检测发现,例1患儿AVPR2基因外显子2上的半合子突变c.650C>T(图1),该突变系新发突变。突变导致AVPR 2 蛋白第217 位脯氨酸被亮氨酸取代,经Mutation-taster 及Polyphen 2 软件预测该突变极可能致病(致病性相关分数分别为0.93和0.99)。由于例1父母拒绝行基因检测,未能证实其是否为该突变携带者。例2患儿AVPR2基因外显子1及外显子2缺失(图2),该突变系半合子突变,国内外均未报道。其母亲系该突变携带者,其父亲AVPR2基因未见异常突变。
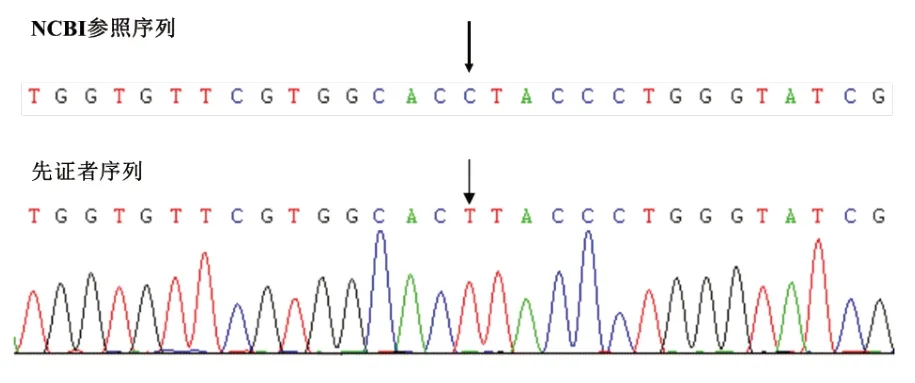
图1 例1 患儿AVPR2 基因测序分析结果(外显子2 c.650C>T)
2 讨论
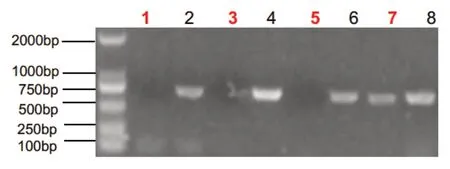
图2 例2 患儿AVPR2 基因外显子1-2 DNA 扩增电泳结果
CNDI发病率低,约占所有尿崩症患者的10%。患儿生后即有尿液浓缩功能障碍,一般生后1 周出现临床表现,如烦躁、喂养困难、体质量下降,出现皮肤干燥、皮肤弹性差、眼球凹陷等脱水表现,间断高热,便秘等[1-2]。但CNDI多起病隐匿,早期可仅表现为多饮、多尿,未受家长重视。国内小样本研究表明,国内尿崩症患儿确诊时的年龄2~15 岁,而国外患儿确诊年龄相对较早,这可能与家长的重视程度及基因检测技术普及程度相关[3-4]。日本学者通过问卷调查收集143例CNDI患儿,发现平均确诊年龄1.2岁,其中69%的患儿于生后第1 年内确诊[2]。本组2 例患儿均于学龄前期就诊,通过病史回顾发现患儿于1 岁左右已出现多饮多尿症状。
通过典型临床表现及实验室检查,即可诊断肾性尿崩症,但CNDI 仍需基因检测以明确诊断。研究表明,90%以上的CNDI 是由AVPR 2基因突变所致。AVPR 2基因定位于染色体Xq 28,基因序列长度大约2.2 kb,包括上游的200 bp,其中包括3个外显子以及3’端非翻译序列,编码1 个含有371 个氨基酸的7 次跨膜蛋白。自1992年发现AVPR2基因突变致CNDI以来,HGMD 数据库记载的基因变异多达286 种。根据AVPR2基因突变类型不同,可将其分为错义突变、无义突变、剪接突变、小片段缺失或插入、大片段缺失或插入、复杂重组等,其中错义突变多见,约占所有突变的48%~52.3%[2,5]。
以“肾性尿崩症、精氨酸加压素受体2或AVPR2”为关键词检索中国期刊全文数据库(CNKI)及万方数据库,以“Nephrogenic Diabetes Insipidus,AVPR2,Chinese”为关键词检索PubMed 数据库,均从建库至2017 年9 月。共检出16 篇有关AVPR 2基因突变的文献。结合本组2例,目前国内已报道的AVPR2基因突变共有56例,39种AVPR2基因突变类型,包括错义突变、无义突变、小片段缺失或插入、大片段缺失或插入、剪接位点异常,其中错义突变约占69.6%[3-4,6-21]。本组患儿中,例1 为错义突变,例2 为大片段缺失,国内外均未报道。新的突变位点不断发现能丰富CNDI疾病谱,为深入研究提供帮助。也有学者根据CNDI发病机制,将AVPR2基因变异分为以下4种:①突变干扰基因转录,使mRNA合成受阻或蛋白翻译异常;②突变导致受体蛋白折叠异常,使其滞留于内质网中,随之被降解;③突变导致精氨酸与受体蛋白结合异常,或受体蛋白与Gs蛋白偶联异常;④突变使胞内的信号转导障碍[1]。研究表明,一种基因突变可能影响受体蛋白的多种功能,从而出现不止一种基因突变类型。例1系c.650C>T的半合子突变,导致受体蛋白第217 位脯氨酸被亮氨酸取代,亮氨酸可与第220 位甘氨酸、第221 位异亮氨酸形成新的氢键,使得受体蛋白结构功能异常,出现尿崩症临床表现。曾有报道1例AVPR2基因突变c.649C>A,导致氨基酸序列改变P217T[22]。该位点与例1患儿突变位点位于同一密码子,两者均造成氨基酸序列改变,且软件预测两者均有强致病性。例2系外显子1及外显子2缺失。有研究者认为,AVPR 2 蛋白的正确转运、组装、成熟至少需要341位氨基酸,大片段丢失必然导致翻译蛋白截短,可能使AVPR 2 受体蛋白滞留于内质网,干扰AVP 与受体蛋白及受体蛋白与Gs蛋白结合,胞内信号转导异常,从而影响肾远曲小管的尿液浓缩功能[6]。
目前CNDI 无特效治疗措施,临床上以减少尿量等对症治疗为主,以防止脱水、肾功能损害等并发症。常见措施包括低盐饮食,适量饮水,非甾体类消炎药、噻嗪类利尿剂及去氨加压素(DDAVP)等[1-2,23]。研究表明,通过以上措施可使患者尿量减少30%~70%。目前CNDI治疗的首选药物为氢氯噻嗪。日本学者通过回顾日本境内CNDI患儿的临床资料发现,约84%(120/143)的患儿应用氢氯噻嗪治疗,其中92%的患者尿量可明显减少[2];而对疗效欠佳者或为早期控制症状,临床上多采用氢氯噻嗪联合其他药物的方法。本组2例患儿均给予氢氯噻嗪联合吲哚美辛治疗,例2患儿饮水量及尿量明显减少;但例1患儿仅夜尿明显减少,饮水量及尿量仍多,这可能是由于不同基因位点突变与其临床表型、治疗效果有一定的相关性。众所周知,DDAVP主要用于治疗中枢性尿崩症,但有研究发现大剂量DDAVP对p.A37P、p.D85N、p.R104C及p.Y128S四种基因变异类型的CNDI患儿同样有较好的疗效[2]。而一旦延误治疗CNDI患儿可出现智力异常、巨大膀胱、输尿管扩张、肾盂积水等。研究发现,肾性尿崩症患者中泌尿系并发症发生率约为42%,其中CNDI患者中肾盂积水及输尿管积水最常见[2]。本组2例患儿泌尿系B超及肾功能检查均未见明显异常。但尿量增多及利尿剂应用会加重肾脏负担,后期有发生泌尿系疾患的可能,因此必需定期随访泌尿系B超。
综上所述,CNDI起病隐匿,临床少见,延误治疗可出现严重并发症,因此临床上应给予重视。临床规范的试验及基因检测技术能帮助明确诊断,提供新的治疗措施,做到早发现早治疗,对改善患者的生活质量有很大帮助。
猜你喜欢
家庭医药(2021年15期)2021-12-01 22:46:06
家庭医药(2021年8期)2021-07-28 23:18:42
透析与人工器官(2020年1期)2020-11-16 01:42:28
养生大世界(2019年12期)2019-12-11 10:08:01
家庭科学·新健康(2018年8期)2018-10-30 10:23:20
中国实用医药(2016年19期)2016-08-05 22:41:35
恋爱婚姻家庭·养生版(2015年12期)2015-05-14 20:46:25
中国药业(2014年19期)2014-05-17 03:12:16
保健与生活(2014年3期)2014-04-29 00:44:03
疑难病杂志(2014年12期)2014-04-16 05:19:28
