
尿中241Am的分析方法研究进展
2020-03-05 09:36高嘉敏文富平娄海林
核化学与放射化学 2020年1期
高嘉敏,文富平,娄海林
中国原子能科学研究院 辐射安全研究所,北京 102413
241Am是锕系核素,半衰期约为432.7 a,主要衰变方式为α衰变,α射线能量主要有5.49 MeV(85.2%)和5.44 MeV(12.8%),也可释放59.54 keV的γ射线。Am在水溶液中能以4种价态存在,即Am(Ⅲ)、Am(Ⅳ)、Am(Ⅴ)、Am(Ⅵ),其中三价是最稳定的价态,以水合离子Am3+·xH2O的形式存在,Am(Ⅲ)水溶液为粉红或红色[1]。
Am的主要来源是大规模核试验在同温层生成的Am,随着地球转动而沉降至各处的全球性落下灰[2]。由于禁核试验条约的签订,现在核试验已经不再产生新的Am,原本由核试验产生的Am也在逐渐衰变减少,但考虑到同样由核试验产生的241Pu、242Pu等核素衰变会产生Am,因此由核试验产生的Am在短期内并不会发生明显的减少,而是会出现一个峰值阶段,然后再逐渐减少,预计在21世纪中叶达到最大值[3]。
目前在动力堆乏燃料后处理特别是钚循环工艺运行、MOX元件制造、高放废液中241Am的分离及241Am同位素核技术应用等方面均会释放241Am。随着我国核工业的发展,待处理的乏燃料中超铀核素的总质量会不断积累[4]。据预测,每吨辐照燃料中所含有的241Am列入表1[5]。

表1 每吨辐照燃料中所含有的241Am[5]Table 1 Amount of 241Am in one ton spent fuel[5]
核事故会造成局部环境内241Am的严重污染,同时核恐怖事件也逐渐成为人们关注的重心,241Am极易被用于放射性散布装置RDD(radiological dispersion devices),也可能产生局部的大量污染。
241Am具有高毒性,进入人体后会沉积在骨骼、肝脏、肌肉组织中,不易排出[6]。相应在放射源生产、核设施退役、反应堆检修等开放性操作环境中的工作人员有可能会受到241Am产生的内照射。因此为工作人员的身体健康考虑,需要对241Am的职业照射进行常规监测,并依此估算241Am在人体内的积存量,确保工作人员受到的照射不超标。
根据美国能源部、核管会对放射性散布装置RDD事件的研究,241Am是其中高风险的核素,劳伦斯利弗莫尔国家实验室对RDD事件进行模拟,发现其中241Am对总有效剂量当量的贡献很高,超过了其他常见放射性核素(90Sr、137Cs、60Co、192Ir)[7]。因此在发生核事故、核恐怖威胁等情况时,也要求对尿中241Am进行快速应急监测,以便于后续应急响应行动的决策。
由于通常尿样中241Am含量较低,因此测量所需的尿样体积较大、需要浓集,在批量处理大体积尿样及应急监测时需要快速测量。尿样中241Am的测量通常需要经过预处理、分离纯化、制样、测量等流程。对于一般大体积的尿样,需要进行预浓集[8],对于体积较小的应急样品,在满足探测要求的情况下可以省略预浓集流程[9]。测量仪器主要有α谱仪、液体闪烁谱仪(LSC)、γ谱仪、电感耦合等离子体质谱(ICP-MS)、加速器质谱(AMS)等,一般的常规测量主要使用α谱仪,快速测量时为达到较快的测量时间通常推荐采用ICP-MS、AMS等质谱方法,同时常见的LSC和γ谱仪也能达到相对较短的测量时间,但和前两类仪器相比,其探测限和分辨率不高。
1 化学分离方法
1.1 样品预处理
收集到的尿样一般先加入浓硝酸或盐酸处理,并放入冰箱保存[10]。由于锕系元素极易吸附在容器壁上,取样后对样品进行酸化是非常重要的步骤。Lovett等[11]研究加酸(pH=1)与不加酸的海水样品中241Am的吸附情况发现,不加酸的样品在2周后损失了约50%的Am(置于25 L聚乙烯容器中)。
在进行化学处理之前,要加入示踪剂以便于计算最终回收率。Am元素常见两种同位素为241Am和243Am,如果样品中不含有或含有极少量243Am,则可使用243Am作为示踪剂。若无法确定样品中是否含有243Am或本身含有243Am时,那么便可以使用148Gd来确定241Am的回收率,148Gd是释放低能α射线的稀土元素,在能谱上能较好地与Am同位素分离开来,且Gd3+的化学性质和Am3+非常相似,在化学处理中的表现与Am类似,故可以用于估计Am的回收率[12]。另外也可以使用Eu同位素来进行标记,但Eu是γ核素,故不便在使用α活度测量仪器时使用。
在样品预处理时,先加入硝酸和过氧化氢并加热干燥,硝酸可以破坏尿样中的有机物,并有助于将胶体和颗粒物中的241Am转变为可溶状态,过氧化氢则可以氧化有机物。之后可采用共沉淀法对尿样中的241Am进行分离浓集,再用离心分离、过滤、灰化等其他方式进行处理,以便后续化学分析或仪器测量。
尿样体积较大或241Am含量较低时,多采用共沉淀法进行预浓集,常用的载体有磷酸钙、草酸钙、Fe/Ti的氢氧化物等。可在尿样中加入硝酸钙和草酸盐(或磷酸盐)使尿样产生沉淀,多次加硝酸等处理直至得到灰白色沉淀,最后将处理后的沉淀溶入酸性溶液中。在快速测量时,可省略较为复杂的共沉淀步骤,直接在尿样中加入浓硝酸/盐酸、亚硝酸盐等进行酸化并调整Pu等其他元素的价态,还可以使用预过滤树脂过滤去除样品中的有机物,然后直接过柱。
总体来说,样品在过柱前需要进行酸化调节pH、价态调整(Pu等其他锕系元素)、去除有机物和干扰核素等预处理操作,但需要根据样品体积、包含核素水平、目标测量时间、测量性能要求等多方面考虑选择必要操作。
1.2 分离纯化方法
分离241Am的方法主要有萃取色谱法、离子交换法、溶剂萃取法等,对于大部分测量方法,如α谱仪、LSC、ICP-MS、AMS等均需要进行分离纯化,仅在后续制样步骤有所区别;但使用γ谱仪时,若测量结果能够满足要求,可以不进行本步骤中的化学处理。
1.2.1萃取色谱法 萃取色谱法结合了溶剂萃取和色谱分离法的优点,是现在最为常用的分离方法,目前使用较为广泛的是已经商品化的有机固定相萃取树脂,主要是美国Eichrom公司研发的DGA树脂、TRU树脂以及ACTINIDE树脂,我国20世纪70年代曾使用HDEHP-Kel-F萃取色谱柱分离方法[13]。DGA树脂有两种:一种是常规型,萃取材料为N,N,N′,N′-四正辛基二乙醇酰胺(N,N,N′,N′-tetra-n-octyldiglycolamide,TODGA);一种是支链型,萃取材料为N,N,N′,N′-四-2-乙基己基二乙醇酰胺(N,N,N′,N′-tetrakis-2-ethylhexyldiglycolamide, TEHDGA)[14]。两种DGA树脂对Am的吸附性能基本相似,只是支链型DGA树脂在相同浓度的硝酸/盐酸下吸附性能略低于常规型DGA,另外在高浓度硝酸下支链型DGA树脂对Pu的吸附性能稍好于常规型。TRU树脂的萃取材料为溶解于磷酸三正丁酯(tri-n-butyl phosphate, TBP)中的辛基(苯基)-N,N-二异丁基胺甲酰基甲基氧化膦(octylphenyl-N,N-di-isobutyl carbamoylphosphine oxide,CMPO,图1),工作容量为每毫升树脂2 mg Am,或每2 mL预填充柱4 mg Am,这个值是树脂理论最大承载能力的20%。TRU树脂吸附四价和六价超铀核素的性能较好,吸附Am(Ⅲ)时需要在大于0.5 mol/L的硝酸环境中,盐酸环境中吸附镅的效果很不好[15]。ACTINIDE树脂基于Dipex萃取剂,对锕系核素有很好的吸附性能。对于浓度小于1 mol/L的酸,ACTINIDE树脂对锕系核素的吸附性能比TRU树脂高,因此ACTINIDE树脂对预浓集大体积液体样品中的锕系核素非常有用,在进行液态流出物监测时,批量使用该树脂然后直接使用液闪测量是可行的快速方法[16]。ACTINIDE树脂对Am(Ⅲ)的吸附很好,但其洗脱没有DGA树脂容易。TRU树脂对Am(Ⅲ)的吸附也很好,使用0.5~5 mol/L HNO3的保留因子为100,但DGA树脂对Am(Ⅲ)的保留因子是TRU树脂的30~500倍[14]。两种DGA树脂在Am的分析方面相较其他树脂有更好的应用前景,它们能够选择性分离Am(Ⅲ),在5 mol/L HNO3或HCl环境中Am(Ⅲ)能够很好地被DGA树脂吸附,然后再用0.01 mol/L HNO3或0.5 mol/L HCl洗脱即可,但用0.01 mol/L HNO3解吸时会存在解吸不完全的现象。近年来大部分对241Am的测量均选择了DGA树脂[7,17],也有一部分进行锕系核素联合测量会选择TRU树脂[18-19]。由于两种树脂对其他锕系核素或超铀、镧系核素也有吸附能力,这些核素大部分都是α辐射体,易对测量产生干扰,因此在分析含有多种核素的样品时需要注意去除其他核素。在使用时,有重力过柱和真空箱过柱两种系统。
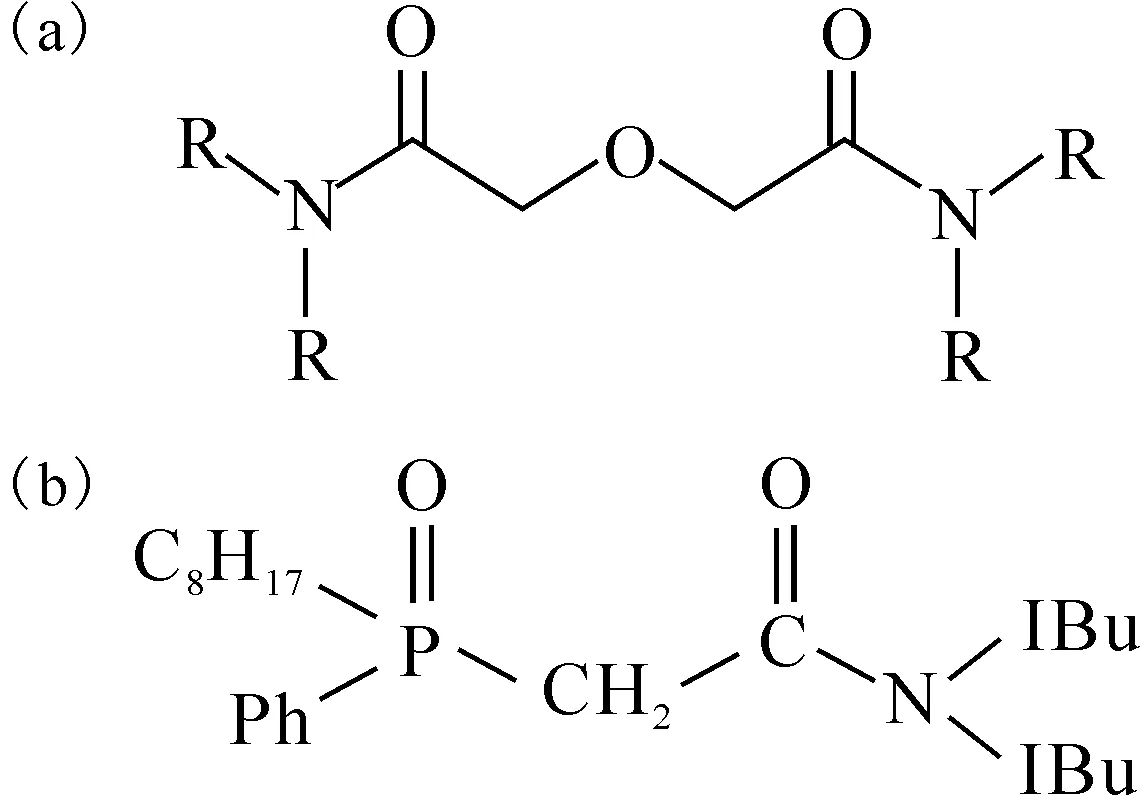
图1 DGA(a)和CMPO(b)结构图Fig.1 Molecular structure of DGA(a) and CMPO(b)
1.2.2离子交换法 阴离子交换柱在浓HCl环境下吸附Pu(Ⅳ)和U(Ⅳ),在浓硝酸环境下吸附Th(Ⅳ)和Pu(Ⅳ),但Am3+在浓硝酸或浓盐酸环境下均不会形成络阴离子,也就不会被吸附在柱子上,因此若使用水溶液环境下的阴离子柱,可以将浓硝酸或浓盐酸淋洗后的洗出液用于241Am分析,后续分析常使用萃取色谱柱。若单独使用阴离子交换柱进行分析,241Am要处于非水溶液环境中才能形成阴离子,常用CH3OH环境,如上柱的载带溶液为1 mol/L HNO3-93% CH3OH(体积分数,下同)溶液,阴离子交换柱先用1 mol/L HNO3-93% CH3OH溶液冲洗去除UO2+、Th4+、Pu4+,用0.1 mol/L HNO3-0.5 mol/L NH4SCN-80%CH3OH溶液冲洗去除镧系元素,上柱后用1.5 mol/L HCl-83%CH3OH洗脱241Am,得到的洗脱液即可直接用于后续制样和测量[20]。常用的阴离子交换柱有Bio-Rad实验室的AG系列树脂、Eichrom的离子交换树脂、Dowexl阴离子交换树脂等,邱咏梅等[21]用国产717强碱型阴离子交换树脂进行实验也得到了较好的回收率。
1.2.3溶剂萃取法 溶剂萃取法在分离高放废液中镅及镧系核素中应用广泛,在实验室放化分析中的应用多见于早期,对于体积较小的样品也常将萃取剂制成萃取色谱柱。常见的萃取体系有二(2-乙基己基)磷酸(HDEHP)、三正辛基氧化膦 (TOPO)、三异辛胺(TIOA)/二甲苯、氯代磷酸双(2,4-二氯苯基)酯 (DDCP)等。Ballestra等[22]对环境中低水平样品用HDEHP萃取并结合硝酸-甲醇介质进行阴离子交换,但由于样品种类、HDEHP也可萃取硝酸的影响,萃取效率不稳定;随后又采用DDCP萃取,消除了溶液中硝酸的影响,先将预处理后的样品通过离子交换柱,蒸干后用硝酸溶解,再用DDCP萃取两次,然后每次用2 mol/L硝酸反萃取两次镅,钍、钚则留在有机相。Dacheux等[12]直接用萃取剂闪烁液混合物萃取水样品中的镅,取有机相脱氧后用α液体闪烁谱仪(PERALS system)测量,萃取闪烁剂有三种,分别为ALPHAEXα(含HDEHP)、THOREXα(含伯胺)或URAEXα(含叔胺三辛胺)。使用TIOA/二甲苯体系萃取,Pu、Po、U和Fe均会萃取进入有机相,而Am会留在水相。Hampson等[23]用TOPO-HNO3体系萃取,并用碳酸铵作反萃取溶液和电沉积液分离生物样品中的锕系核素。溶剂萃取法的样品溶解过程比较复杂,不适用于大批量样品的快速分析测量。
1.2.4影响回收率的因素
(1) 尿素含量
尿样中所含的主要物质是尿素,在使用萃取色谱柱法时,尿素含量高可降低241Am的回收率。Zagyvai等[24]发现尿素含量对洗脱DGA柱上的241Am有影响,原因是尿素和DGA分子结合形成了一种混合物,这种混合物固定241Am的能力很强,使得241Am不易被洗脱下来。如果能快速直接分离241Am的话,可达到80%以上的回收率,如100 mL尿样过DGA柱后直接用0.5 mol/L HCl洗脱,这种方法快速但不是很精确。另一种方法是用65%硝酸过柱,破坏其中的TODGA分子,这种方法较慢但更精确。
(2) HNO3含量
基质作用会影响色谱柱分离过程的回收率,可通过改变负载溶液中硝酸的浓度来提高回收率,当硝酸的浓度从3 mol/L上升到5 mol/L时,241Am的回收率从66%上升到88%,但硝酸浓度继续升至6 mol/L时,241Am回收率又急剧下降至42%,因此在与Horwitz等[14]方法相似的色谱柱分离流程中用5 mol/L的负载溶液241Am回收率最好[7]。
(3) 二乙基三胺五乙酸(DTPA)的影响
辐照工作人员或公众在吸入Am后,若服用促排药物DTPA治疗,会使得尿样中也含有DTPA。DTPA对DGA吸附241Am没有影响[24],对HDEHP萃取241Am有影响,但DTPA对三价镅的络合作用受介质中酸度制约,因此适当控制介质酸度可降低DTPA络合镅的干扰,赵敏等[13]发现在HEDHP-Kel-F柱上,介质pH值在2.0时,20 mg DTPA基本不影响镅的吸附。
(4) 离子的影响
铁离子会影响241Am在ACTINIDE树脂上的吸附,Fe3+的影响很大,Fe2+的影响较小,因此若预计尿样中含有较多Fe3+,可提前加入还原剂如抗坏血酸将其还原为影响较小的Fe2+。与ACTINIDE树脂类似,Fe3+对241Am在TRU上的吸附也有负面影响,而Fe2+几乎没有影响,因此也可在基质中加入还原剂,并用硫氰酸铵作指示剂,其在Fe3+存在时显深红色,仅有Fe2+时则无色。另外,Al3+对TRU吸附241Am的影响较大,Ca2+则几乎没有影响,因此Ca2+、Fe2+可用作共沉淀的载体。使用DGA柱时,Fe3+在盐酸浓度较高时吸附较多,但在HNO3环境下没有吸附,因此可选用硝酸或低浓度盐酸。
1.3 制样方法
分离纯化步骤之后, 还需要制备适应不同测量仪器的样品源。由于α粒子的射程很短,因此利用α谱仪测量时需要将测量样品制成均匀、高纯度的薄源。常用的α谱仪制样方法有蒸发法、微量共沉淀法、电沉积法。
蒸发法是将样品溶液直接在金属盘上加热蒸干,这种方法简便快速,但可能将其他干扰核素一并干燥到平板上,能量分辨率较差。蒸发法的另一个问题是干燥时容易在平板边缘形成环,使得样品不均匀,可以采取添加湿润剂(如四甘醇或5%(质量分数)胰岛素溶液)、冷冻干燥样品或沉淀等方法防止样品损失及不均匀问题[25]。
电沉积法是通用且常见的方法,能量分辨率好,其原理是使镅化合物水解产物在适宜的pH值范围内沉积在不锈钢阴极表面。电沉积液分为两类,有机电沉积液和无机盐类电沉积液。有机电沉积液一般电阻较高,所需的电压也较高,不易直接用于水溶液样品。无机盐电解质溶液中,以微酸性的铵盐溶液最为常见,如硫酸铵、草酸铵、氯化铵等[26]。草酸铵体系需要热水浴加热,在电沉积过程中pH值变化大,需要随时调节;硫酸铵体系不需要加热和调节酸度,对阴极材料损伤小;这两种体系均不需要蒸发电解溶液、不需要为完全电沉积而耗费过长时间,适用于纯溶液的电解沉积。盐酸具有腐蚀性,对阴极材料有较高要求,复杂基质环境样品可使用氯化铵电解沉积,但必须防止氯化铵溶液蒸发过度[27]。综合来说,选用硫酸铵体系沉积241Am的效果最好,操作方便、沉积率高。电沉积液的pH对电沉积率有影响,过低的pH值会增加电极的腐蚀,使源片外观变差、分辨率降低,当pH值为1.8~3.5时,241Am的电沉积率基本不变。电沉积时间一般在100 min左右,电沉积结束后,可将电镀片在500~600 ℃加热300 min,则其可抵抗物理破坏,不会再被洗掉,然后用乙醇和水冲洗源,最后用带酒精的棉花球擦拭[28]。硫酸铵适用于沉积多种锕系核素,Talvitie[29]在pH=2的(NH4)2SO4中电沉积Pu、Th、U和Am效果较好,目前仍有人使用这种方法[26-27];Ballestra等[22]也在pH=3的(NH4)2SO4介质中电沉积镅和锔;硫酸铵沉积120 min时镅的沉积率达90%以上,草酸铵沉积90 min时镅的沉积率达80%以上[30]。喻正伟等[31]利用自制的电刷镀仪,采用pH≈3.5的异丙醇-硝酸电镀液制备大面积241Am放射源,最大沉积率可达到80%。
电沉积法虽使用历史较久、应用较多,但仍存在制备时间较长、沉积较厚、回收率较低等缺点,而微量共沉淀法则可以最小化这些缺点。微量共沉淀法较为快速,回收率高,分辨率可接近一般电沉积样品,且在出现问题时,可以将样品溶解后重新制备,此外,微量共沉淀法不需要复杂的设备,简单的过滤系统即可进行操作。其缺点在于,制备过程中会用到有害的氢氟酸,因滤膜不平而导致的不可重复和不明确的计数几何条件,并且不适于长期保存[32]。微量共沉淀法常用的载体为镧系元素,如Ce、Nd、La等,其比Fe或Zr等较小的离子效果更好。在镧系元素中,Ce因与Nd和La相比具有额外的氧化态而更易被净化,因此在制作241Am样品时,Ce比Nd、La应用略多一些。微沉淀的滤膜使用0.1 μm孔径的聚丙烯滤膜可得到较好的回收率和能量分辨率,但需注意聚丙烯滤膜的熔点较低,要防止其发生卷曲或融化。一般在室温下,沉淀20 min即可得到较好的回收率。
利用LSC测量时将洗脱液与闪烁液混合即可。在能达到要求的情况下,γ谱仪可直接测量样品,不需前处理。
质谱测量时,在pH适宜的情况下,洗脱液可直接用于ICP-MS测量,也可将洗脱液蒸干再溶于10 mL 0.28 mol/L HNO3中[33]。AMS测量时,可先用Fe/Ti的氢氧化物共沉淀241Am,蒸干后的沉淀物与4~5 mg Nb粉混合压入Ti靶即可用于测量[34]。
2 测量方法
241Am的测量方法主要有两大类:一类是放射性测量方法,α谱仪和液闪谱仪可测量其释放的α射线,γ谱仪可测量其释放的γ射线;另一类是非放射性测量方法,主要有质谱方法,其中常用的有电感耦合等离子体质谱(ICP-MS)、加速器质谱(AMS),其他可用方法还有热电离质谱(TIMS)、共振电离质谱(RIMS)、荧光光谱、红外光谱分析法等。
2.1 α谱仪
α谱仪测量是应用最为广泛且灵敏度较高的方法,但由于超铀核素大部分都是α源,因此在使用α谱仪前必须进行化学前处理并制成薄源,去除其他α放射性核素和基质中的干扰物质并减少自吸收,尤其要去除Pu、Th、Ra和Po,因为这些核素的α射线能量与241Am非常接近[35]。根据测量时间的不同,α谱仪测量的探测下限(MDA)范围为0.1~20 mBq/L。
2.2 液体闪烁谱仪
液体闪烁谱仪也可用于241Am的测量,样品前处理的方法与α谱仪的处理流程类似,但只需要得到液体样品再加入闪烁液即可进行测量。Sadi等[7]发现取7.5 mL洗脱液混合12.5 mL闪烁液(Optiphase HiSafe Ⅲ闪烁液)得到的回收率最高,使用普通商用液闪谱仪(Tri-Carb 3180 TR/SL 液闪谱仪)计数10 min得到的探测下限是1.3 Bq/L。
普通商用液闪谱仪主要用于测量β放射源,特别是低能β核素,其用于测量α核素的效果不是很好,能量分辨率较低,FWHM只能达到1 MeV左右,β-γ本底较高,还存在不规则的淬灭作用。考虑到普通β液闪的缺点,McDowell等[36]研制了专门用于测量α核素的液闪谱仪,即光电过滤α液体闪烁谱仪(PERALS spectrometry,photon/electron rejection alpha liquid scintillation spectrometry,商标为ORDELA)。这种谱仪使用电子脉冲形态鉴别(PSD)滤除β和γ脉冲来降低本底,选择最佳的探测器结构和相关的α脉冲电子仪器,并使用液-液萃取闪烁剂。尽管PERALS的能量分辨率不如一些高分辨率α谱仪,但相较β液闪谱仪,其分辨率提高至250 keV以上,探测限也从10~20 min-1提升至0.01 min-1 [37]。Dacheux等[12]使用液-液萃取和PERALS系统测量241Am的探测限为6.8×10-4Bq/L(测量时间为3 d),测量1 d时的探测限为1.5×10-3Bq/L。
2.3 γ谱仪
γ谱仪测量是较为简便直接的方法,其测量的样品甚至可以不需化学处理的过程,这对样品没有破坏性,也可以满足样品后续其他分析测量的需要,但使用时需要修正样品中γ射线的衰减,保证测量结果准确。由于高纯锗探测器的计数效率较低,且为防止样品源对γ射线的自吸收,样品源体积一般较小或做的比较薄,所以γ谱仪的灵敏度通常比较低,一般用于测量0.1~1 Bq/L甚至更高水平的样品。对于水平较高样品的快速测量,可对样品加酸处理,用氢氧化物沉淀,再用草酸盐或氟化物沉淀后直接用γ谱仪测量。Li等[38]在分析130 mL尿样中Am时,不做前处理,20 Bq/L水平下测量30 min、2 Bq/L水平下测量90 min均可达2 Bq/L的探测下限,不确定度分别为9%~13%和10%~43%。想要得到更好的探测下限,可以使用大体积尿样并进行化学处理。
2.4 电感耦合等离子体质谱(ICP-MS)
电感耦合等离子体质谱是测量痕量和超痕量元素及同位素最常用的质谱方法,在测量放射性核素方面也应用越来越广泛,常用于测量长寿命放射性核素,也是测量241Am最快的方法。
与传统的无机固体质谱不同,ICP-MS允许在大气压下操作的离子源中引入样品,并使用标准水溶液进行简单的量化测量。激光消融系统耦合ICP-MS可以直接分析固体样品。在ICP-MS中,样品溶液中的化合物在约6 000~8 000 K下,在电感耦合氩等离子体中分解成原子,高温保证了高电离度(对大部分元素来说大于90%),使其只含有少量的多电荷离子。正离子在大气压下从电感耦合等离子体通过界面进入质谱仪的高真空环境中。然后将提取的离子用四极杆飞行时间质量过滤器或电磁和静电组合扇形区进行分离,最后用离子检测器测量。根据干扰因素和仪器灵敏度的不同,ICP-MS的探测限从10-15~10-8g不等。在低分辨率模式下,双聚焦扇形场(sector field, SF)ICP-MS比传统四级杆型ICP-MS的灵敏度更好。
用ICP-MS测量的主要问题是等重元素(如129Xe对129I)或等重多原子分子(如238U1H和204Pb35Cl对239Pu)的干扰,在双聚焦扇形场ICP-MS中,等重分子可以被分解,但由于使用了较强离子束瞄准会使得灵敏度下降;对于四级杆型ICP-MS,通过选择碰撞室/反应室(DRC)的反应气体可以抑制同重离子的干扰。
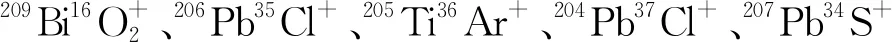
2.5 加速器质谱(AMS)
加速器质谱是非常灵敏的可用于分析中长及长寿命放射性核素的技术,由于其能够抵抗等重分子离子的干扰,并且受基体效应的影响小,所以加速器质谱技术能够简化样品化学前处理过程,并能够带来较高的样品分析产量,减少超低水平生物检测的成本。对于生物检测样品中超痕量重锕系核素的探测来说,加速器质谱非常重要,其他的质谱分析方法(如ICP-MS和TIMS)可能无法达到相同的分析灵敏度和产量要求[39]。在使用AMS测量时,由Cs+溅射样品得到氧化锕阴离子(AnO-),然后射入终端电压约为320 kV的加速器中。在终端,氦被用作剥离器气体来瓦解剥离分子离子并收集正锕离子。在高能侧,通过一列由2个高能磁铁和静电分析器组合的装置来筛选出An3+。最后An3+由一个专用的低噪音气体电离探测器计数,该气体电离室有一个30 nm厚的SiN窗来阻挡其他相似荷质比离子的干扰。Dai等[34]使用瑞典皇家理工学院的紧凑型AMS测量尿样中Am和Cm,使用混合氧化Fe/Ti制作样品靶,测量探测限约为0.1~0.2 fg/L(0.01~0.03 mBq/L),好于α谱仪能够达到的约0.1 mBq/L(约相当于0.6 fg/L)的最小探测限(在低本底条件和长达几周的足够的计数时间下)。
2.6 镅及其他锕系核素联合测量方法
为快速得到测量结果,也常将镅与其他锕系核素及90Sr进行一次通过式的联合测量。联合测量的前处理流程与单独处理241Am类似,但需要在上柱前进行价态调整,并串联多个色谱柱。用于测量超铀核素的树脂有UTEVA、TEVA、TRU、DGA、ACTINIDE等,测量90Sr可用SR树脂、AnaLig90Sr树脂、离子交换树脂等,可根据测量需求将这些树脂组合使用,如TEVA+TRU+SR树脂、TEVA+DGA+SR树脂等,过柱得到的洗脱液再根据核素的不同分别制样,最后用相应的仪器测量[8,10,19,40-42]。
3 总 结
本文总结了尿中241Am的各种分析和测量方法,各种方法的简明对比列于表2。根据处理的时间要求,分为快速测量和常规测量两种。快速测量可采取简便的化学处理和快速测量仪器例如质谱仪。常规处理及要求精度较高或241Am水平较低的情况,可采取较为复杂的化学处理及长时间的计数测量。对于尿样体积较小(≈10 mL)的情况可直接进行分离纯化,尿样体积较大(>100 mL)则需要进行共沉淀进行浓集处理。萃取色谱柱法结合了溶剂萃取和色谱法的优点,是较为常用的分离方法,但国外商用树脂的价格较高,仍需研发使用流程简便的萃取树脂。α谱仪是能量分辨率及灵敏度均较好、且应用广泛的测量方法,是常规测量的重要仪器,但近年来质谱法迅速发展,也逐渐成为快速、低水平测量的较好方法。目前国内尚未建立分析尿中241Am的标准方法,但有食品中241Am的分析方法可供参考,同时也可参照国外的方法进行分析。总的来说,测量尿中241Am的基本方法已经建立,但仍需在以下几个方面有更好的发展:(1) 由于尿样中241Am的含量通常较低,因此发展超痕量241Am的分析测量手段仍是目前较为重要的方向;(2) 尿液累积取样时间一般在48 h以上,样品体积相对较大,提高大体积样品测量的回收率也是需要关注的问题;(3) 对工作人员进行常规测量时会面临大批量大体积测量的问题,因此在快速分析测量方法的基础上推进自动化测量系统的建立应是后续研究及应用的重点。
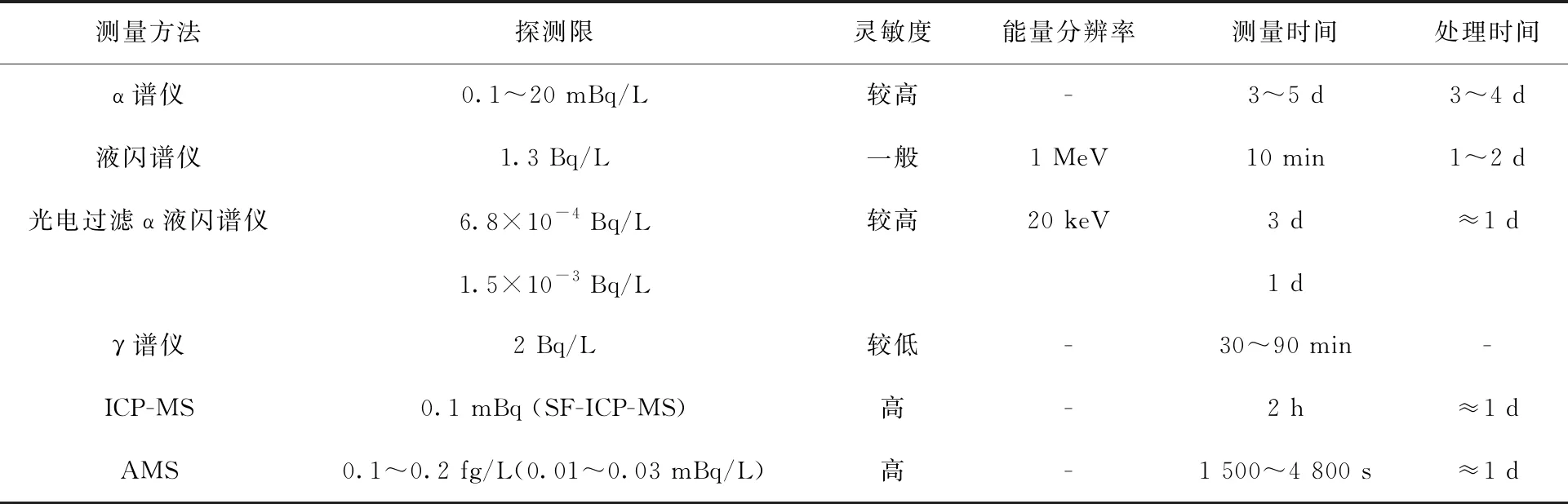
表2 不同测量方法对比Table 2 Comparison of different measurement methods
猜你喜欢
中国伤残医学(2022年14期)2022-12-23
科技创新与品牌(2022年4期)2022-05-08
现代仪器与医疗(2022年1期)2022-04-19
波谱学杂志(2022年1期)2022-03-15
家庭科学·新健康(2022年2期)2022-03-07
科学技术与工程(2021年31期)2021-11-23
国际放射医学核医学杂志(2021年10期)2021-02-28
福建医科大学学报(2020年2期)2020-06-10
空间科学学报(2020年2期)2020-04-01
环球时报(2016-05-18)2016-05-18
