
乏燃料后处理中的辐射化学问题Ⅱ.水溶液和稀释剂的辐射分解
2020-03-05 09:36:14翁汉钦叶国安林铭章
核化学与放射化学 2020年1期
汤 嘉,翁汉钦,何 辉,叶国安,林铭章,3,*
1.中国科学技术大学 物理学院 工程与应用物理系,安徽 合肥 230026;2.中国原子能科学研究院 放射化学研究所,北京 102413;3.中国科学院 核能安全技术研究所,安徽 合肥 230031
自1952年12月美国实现增值堆1号(EBR-1)首次利用核能发电以来,世界核电已有60多年的发展历史。2014年,核电站向全球供应2 441 TWh的电能,这占到当年全球发电量的10%,而中国为了迎合能源结构改革,将大力发展核电。发展迅猛的核电行业带来了经济利益的同时,也随之产生大量的乏燃料。乏燃料能否得到安全有效的处理、处置关系着核电的发展。为了实现核能的可持续发展战略,我国采取对乏燃料进行闭式循环的战略,即对乏燃料进行后处理,回收铀、钚,并通过再循环加以利用,以提高核燃料的利用率,减少放射性产物的毒素和体积[1]。
关于乏燃料后处理的元素分离,已发展出多种商用和实验室阶段的后处理流程,如PUREX流程[2]、UREX流程[3]、COEX流程[4]、CSEX流程[5]、SREX流程[6]、FPEX流程[7]、TRUEX流程[8]、DIAMEX流程[9]、ARTIST流程[10]、TALSPEAK流程[11]、GANEX流程[12]。由于乏燃料具有放射性的特殊性,这些湿法流程中水相和有机相不可避免会受到238Pu、241Am等α辐射源产生的α辐射以及裂片核素如90Sr、137Cs产生的β和γ辐射。该过程中产生的辐射使得水相和有机相的物质发生化学反应。简而言之,在乏燃料后处理中需要关注辐射化学的主要原因在于:(1) 辐射分解降低了配体浓度从而导致萃取效率降低;(2) 辐解产物往往是新的络合剂从而导致分离因子降低;(3) 辐解产物经常是亲水性的络合剂,因而对反萃不利;(4) 辐射分解产生的中间活性粒种可能改变金属的价态。
本综述系列Ⅰ[13]已经对磷酸三丁酯(TBP)及多种新型的萃取剂的辐射稳定性进行了介绍和讨论。本文作为系列Ⅱ,将主要从硝酸辐解、锕系水溶液化学、稀释剂的辐射稳定性以及乏燃料的氧化溶解四个方面介绍,并简单解释辐解的机理。
1 硝酸的辐解
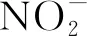
研究硝酸辐解的途径主要包括稳态辐解和脉冲辐解两种方法。随着技术的进步以及研究的深入,时间尺度更小的脉冲辐解装置被研制出来[22],为硝酸辐解机理进一步的研究提供了基础条件。
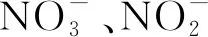


(1)
(2)
(3)
(4)
(5)
(6)
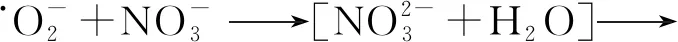
(7)
Garaix等[23]通过13.5 MeV的α辐射对含有NaNO3和HNO3的水溶液进行研究,进一步研究H2O2辐射化学产额,并给出了H2O2辐射化学产额(G(H2O2))的经验公式(式(8)—(10))。
(8)
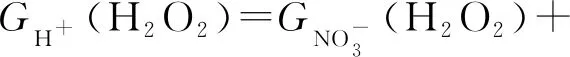
(9)
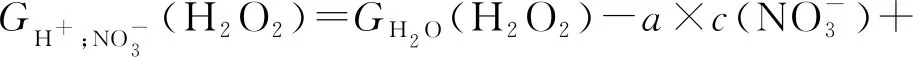
(10)

(11)
加入该机理反应进行模拟后,并在反应速率常数为1.0×1013dm3/(mol·s)时,对于硝酸影响水辐解的G(H2)的模拟结果与文献[25-34]实验数据一致。
Liu等[20]以238Pu作为辐射内源探究了水溶液硝酸的辐解过程,并给出了HNO2产生和消耗的关键方程式(12)—(17)。
(12)
(13)
(14)
(15)
(16)
(17)
式(12)—(17)表明·NO2、·NO和H2O2是HNO2产生的关键中间产物。
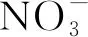
(18)
GH+(HNO2)=B×(1-exp(-g×c(H+)))
(19)
(20)
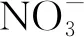

(21)
(22)
(23)
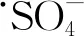
(24)
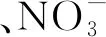
目前对·NO3生成机理的研究已经比较全面[18,46],有学者[49-52]对·NO3的消耗也开展了一定的工作,综合认为·NO3有如下辐解机理(式(25)—(32)),Garaix等[53]通过ELYSE皮秒脉冲辐解装置测定了·NO3辐解过程的化学反应速率常数(k)。
(25)
k=4.0×106L/(mol·s)
(26)
k=4.4×109L/(mol·s)
(27)
k=2.0×108L/(mol·s)
(28)
k=1.1×109L/(mol·s)
(29)
k=3.0×102L/(mol·s)
(30)
k=1.0×1010L/(mol·s)
(31)
k=3.0×109L/(mol·s)
(32)
k=7.1×106L/(mol·s)
Garaix等[53]还通过ELYSE皮秒脉冲辐解装置探究酸度和肼对·NO3辐解的影响,结果表明:在中性无肼条件下,辐解产生的·NO3主要与四种物质发生反应,在单次脉冲为38 Gy的辐射条件下,·NO3主要由·NO2(式(28))、·NO3(式(26))、·OH(式(30))和H2O(式(29))消耗。在纯7 mol/L HNO3溶液、单次脉冲为38 Gy的辐射体系下,·NO3主要和两种物质反应:·NO2(式(28))和HNO2(式(27)),其中80%的·NO3由·NO2消耗,15%的·NO3由HNO2消耗。
由于实际后处理过程中,会引用氧化性物质如N2H4等抑制HNO2的产生,因此Garaix等[53]还研究了7 mol/L HNO3溶液加入N2H4后的·NO3的产额和消耗,发现N2H4的存在可以极大加速·NO3的消耗,机理在于式(33)[54-55]:
(33)
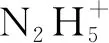
(34)
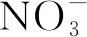
(35)
(36)
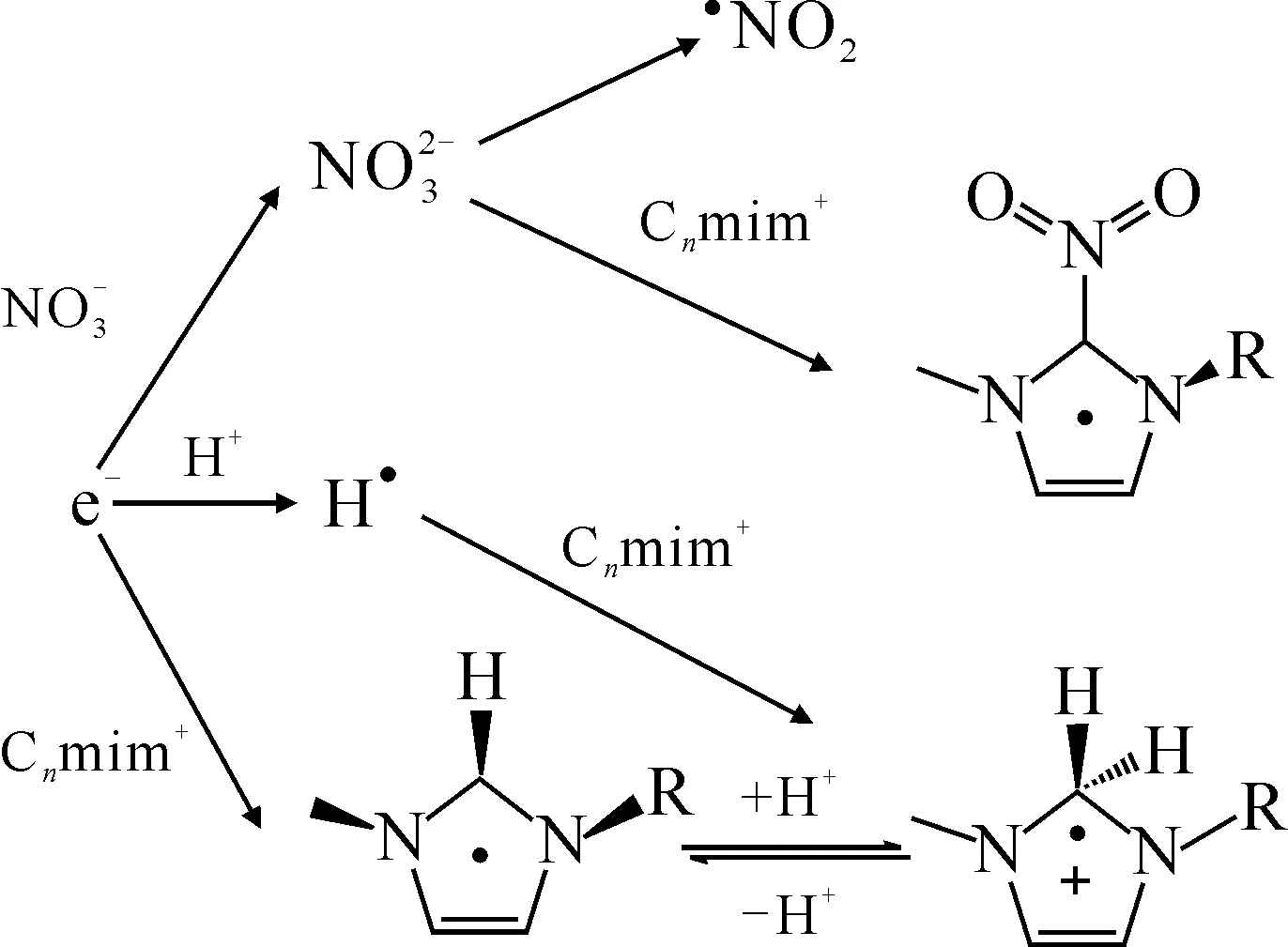
图1 离子液体(阳离子为Cnmim+)辐解体系中硝酸清除e-的机理[56]Fig.1 Mechanism of removal of e- by nitric acid in irradiated ionic liquids(cations: Cnmim+) [56]
2 锕系水溶液的辐射化学
在后处理流程中,锕系元素的价态一定程度上决定了萃取分离的效率。而在电离辐照条件下,硝酸水溶液会产生还原性和氧化性的自由基等产物,从而影响金属离子价态的变化。因此研究锕系水溶液辐射化学有实际指导意义。
Mincher等[58-59]比较了γ辐解条件下硝酸体系中Am(Ⅵ)和Np(Ⅴ)的氧化还原反应。Am(Ⅵ)通过NaBiO3氧化Am(Ⅲ)得到。对Am(Ⅵ)的硝酸溶液进行γ辐解,在0.3 kGy的累积辐射过程中,间断性使用紫外/可见光谱采集数据,分析发现Am(Ⅵ)和Am(Ⅲ)的浓度随着辐照剂量的增加逐渐降低,Am(Ⅴ)的浓度则随辐照剂量的累积上升逐渐增加,这表明Am(Ⅵ)最终转化为Am(Ⅴ)。Am(Ⅵ)转化为Am(Ⅴ)的原因可能是由于持续的辐解,使得部分HNO3和H2O生成HNO2和一些还原性的自由基,这一推断和Grimes等[60]的实验论证不谋而合。Grimes等[60]探究了Am(Ⅵ)在HNO3水溶液中自动还原的动力学,给出了Am(Ⅵ)还原的机理(如式(37)—(38))。
(37)
(38)
Mincher等[59]对Np(Ⅴ)的硝酸水溶液进行辐照时发现:一段时间后,溶液中产生了Np(Ⅵ)的吸收峰,这一现象可解释为Np(Ⅴ)与水中氧化型自由基发生氧化反应。但在持续的辐照后,Np(Ⅵ)的浓度却反而减少,即Np(Ⅵ)被重新还原成为Np(Ⅴ),这与辐射条件下Am(Ⅵ)转化为Am(Ⅴ)的原因可能类似。Np(Ⅴ)被氧化成Np(Ⅵ)后又进一步还原的机理总结如式(39)—(46)。
(39)
(40)
(41)
(42)
(43)
(44)
(45)
(46)
在后处理PUREX流程的实际应用中,TBP萃取剂对Np(Ⅴ)的萃取性能较差,而对Np(Ⅳ)和Np(Ⅵ)选择性高,但Np(Ⅳ)会影响Pu(Ⅳ)的价态,所以人们希望使Np更多以Np(Ⅵ)形式存在。Precek等[61]希望通过引入一些物质来满足这一要求。通过在含Np溶液中加入V(Ⅴ),以期发生反应(47)并清除部分HNO2。
(47)
同时加入甲基脲(MU),以期清除HNO2,见图2所示,而这有利于Np(Ⅴ)氧化成Np(Ⅵ)(式(48))。
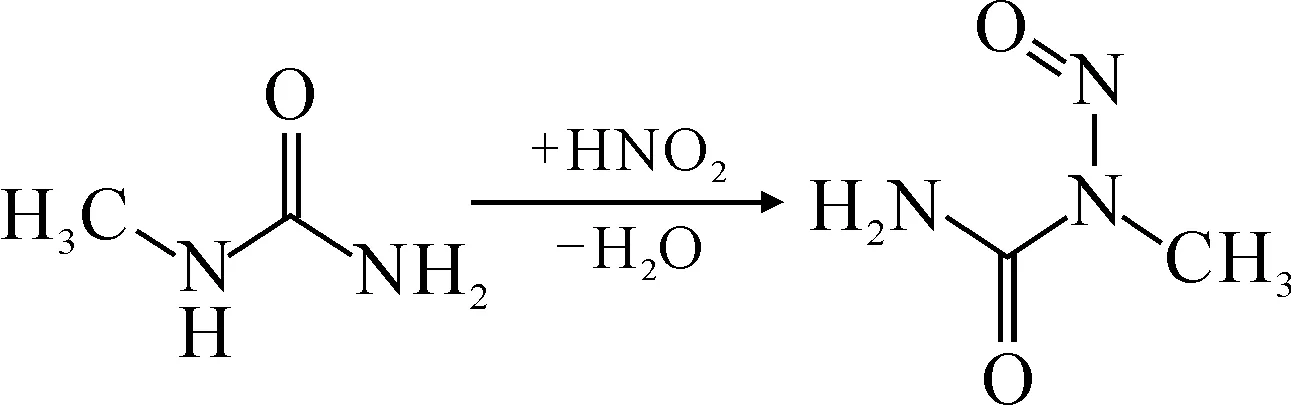
图2 MU与HNO2的反应[61]Fig.2 Reaction between MU and HNO2[61]
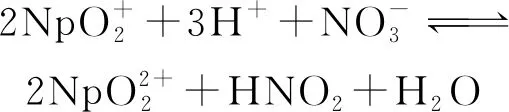
(48)
Precek等[61]实验结果发现:V对Np价态的影响不是很明显,虽然用紫外光谱(UV光谱)检测到了V(Ⅳ),但实验证明V(Ⅴ)并不是被HNO2直接还原,更有可能是被辐解产物中的自由基还原。在MU的浓度达到50 mmol/L以上时,通过增加MU可以促进Np(Ⅵ)浓度的提升,但是从数据上看,提升的程度也不够明显。由UV光谱分析得到:在60 kGy的累积辐射剂量时,90%的Np(Ⅵ)转化成Np(Ⅳ)的形式存在。这对Np的提取有利,但对Pu的萃取却不利。因为Np(Ⅳ)具有还原性,会使Pu(Ⅳ)还原成为Pu(Ⅲ),PUREX流程中的萃取剂TBP对Pu(Ⅲ)的分配系数并不高。

(49)
这使得低硝酸浓度Am(Ⅲ)体系下的水溶液的G(HNO2)较小。而对于低硝酸浓度Pu体系下的水溶液而言,主要有两点原因使得G(H2O2)较高:
(1) Pu(Ⅳ)本身会与H2O2反应(式(50))。
(50)
(2) Pu的价态变化在G(HNO2)上也起到了关键性的作用,Pu在水溶液中会发生反应(51)—(56)。
(51)
(52)
(53)
(54)
(55)
(56)
在Pu(Ⅳ)转变成为Pu(Ⅲ)后,可发生反应(57)。
(57)
Pu(Ⅲ)转化成为Pu(Ⅳ)后又会进行上述反应,使得Pu(Ⅳ)充当着催化剂的角色。这一步反应产生的·NO2会进一步促进HNO2的生成(反应(58)—(59))。
(58)
(59)
这与Liu等[20]认为Pu(Ⅳ)作为一种催化剂,能增加G(HNO2)的观点一致,反应机理如式(60):
(60)
当浓度升高时,G(HNO2)主要还是来自于对硝酸的辐解,这时Pu价态对G(HNO2)的影响逐渐变小,以至于G(HNO2)主要来自于Pu和Am对HNO3的α辐解。
由于H2的易燃性,H2的产生在后处理流程中也是一个需要被考虑的问题。Gregson等[63]对含有Pu(Ⅳ)和Am(Ⅲ)的硝酸溶液的α自辐解产氢进行了一定的探究,发现在硝酸浓度增加时,对于含有Pu或Am的α自辐解体系,G(H2)反而下降。硝酸体系产生H2的机理如式(61)—(64)。
(61)
(62)
(63)
(64)
70%的G(H2)来自于上述反应(64)。在体系为弱酸性时,溶液中的H+会和e-的前体及e-发生反应(65)—(66):
(65)
(66)
这会促进H2的产生。但当硝酸浓度增加时,按照Horne等[24]的理论,会同时发生反应(11)、(67)—(69):
(67)
(68)
(69)
使得H2的浓度降低。而Pu(Ⅳ)和Am(Ⅲ)在弱酸体系中由于自身具有电子清除剂的作用也能起到抑制G(H2)的能力(式(70)—(71))。
(70)
(71)
但硝酸浓度较高时,Pu(Ⅳ)和Am(Ⅲ)影响G(H2)的能力将由于反应(11)的增强而相对减弱。
3 稀释剂的辐解
由于萃取剂的粘度大,密度与水比较接近,直接用萃取剂作为有机相,容易产生第三相,降低萃取性能。为了解决这一问题,必须采用稀释剂。
后处理中常见的稀释剂主要以正十二烷(NDD)和煤油为主。随着后处理的进一步研究,一批新的萃取体系被研究出来,如以正辛醇为稀释剂的6,6′-双-(5,6-二烷基-1,2,4-三嗪-3-基)-2,2′-联吡啶(BTBP)和1,3-双(4-吡啶基)丙烷(BPP)类萃取体系以及以环己酮为稀释剂的BTBP萃取体系等。离子液体ILs具有优良的物理化学性能,比如提供用于离子结合的粒子、较低的挥发性、高电导率、高的极性和极高的金属离子溶解度,在实验室范围作为有机膦类、酰胺类和含氮杂环类萃取剂的稀释剂也已经开展了较多的研究[64-67]。但基于国内已有两篇描述较为详细的综述介绍近年来离子液体的辐射效应研究[68-69],本文主要介绍近十年来已经应用的烷烃类稀释剂的辐解效应研究。
Mincher等[70]对直链烷烃类的稀释剂的辐解已经进行了较为详细的综述。直链烷烃在辐解过程主要会生成以下物质(式(72))。

(72)
在辐解过程最先发生反应(73)—(75)。
(73)
(74)
(75)
由于在直链烷烃的辐解过程中只有小于10%的辐解产物是来自于直接辐解,剩余部分主要通过电离过程产生[71]。上述反应是已知探测尺度下直链烷烃在辐照后最先诱发的反应,对直链烷烃的辐解有着重要的影响。但由于时间尺度较短,实验测定较为困难。Kondoh等[72-73]首次使用飞秒级脉冲辐解装置对NDD的辐解过程中发生的上述反应进行了探究,研究表明辐照产生的RH·+*只有7 ps的寿命。在7 ps内,73%的RH·+*会发生退激反应(反应(74)),并与电子发生重组(反应(75)),剩余的27%的RH·+*直接与电子发生重组。最终RH·+*形成自由离子、RH*、RH**等形态,并发生一系列电离反应。
4 乏燃料的氧化溶解
为了评估地下水与乏燃料潜在接触后地质处置库的安全性,有必要在处置多年后确定乏燃料的溶解特性。因为未“燃烧”充分的乏燃料会释放出α粒子,在水中引起H2O2等氧化性物质浓度的累积,从而会使得固态的乏燃料的价态发生变化,转化成为可溶于水的价态,导致在水溶液中释放更多的α粒子,这一过程对后处理处置十分不利。研究乏燃料在水中的溶解是一个有实际意义的课题。近十年内已有较为广泛的研究[74-76]。图3描述了UO2溶解过程的反应机理[77]。

1—5——反应编号图3 α辐照条件下UO2的溶解机理[77]Fig.3 Dissolution mechanism of UO2 under α-irradiation conditions[77]
Liu等[78]对含有放射性的UO2的封闭体系进行模拟研究,H2O2、O2和H2均会对UO2的溶解产生影响。在封闭的自辐解体系里,O2和H2会因为水的辐解产生并最终到达稳定的浓度。O2会增加溶解速率,而H2的累积则会抑制这一过程直到一个可以忽略的水平。所以UO2的溶解速率主要取决于H2O2的生成速率。
H2O2的生成途径之一为式(76)。
(76)
Shilov等[79]认为该过程中O—O键的结合会释放出能量,一部分给溶剂水分子,一部分会使得H2O2处于激发态,因而以H2O2作为反应物的化学反应更容易达到反应阈能,使得溶液中因辐解产生的H2O2比外加的H2O2与体系内的物质反应速率更快。
对后处理地质处置过程中UO2与辐解的氧化产物如H2O2的接触已经开展广泛研究,但至今均相NpO2和PuO2的放射性溶解动力学的实验在公开文献中却基本上没有。Pehrman等[80]探究了含有NpO2和PuO2的H2O2水溶液消耗的动力学,并将它们的行为与UO2和H2O2的行为进行了比较。实验结果发现,在H2O2存在的条件下,锕系元素的固体氧化物的溶解速率大小分别为UO2>NpO2>PuO2。而溶液中无H2O2时,NpO2和PuO2的溶解速率分别下降90%和85%。进一步证明了H2O2在地质处置中对氧化溶解十分不利。
Odorowski等[81]进一步探究了MOX燃料在有氧和无氧的碳酸盐水溶液中长期(一年)辐照的氧化溶解过程和溶解的锕系元素的形态。相比于PuO2,UO2会优先氧化溶解。这与Pehrman等[80]的结论保持一致:释放的U元素主要以溶解度较高的碳酸盐络合物的形式存在,而Pu和Am主要富集在容器TiO2的壁面上;12%的Am以胶体的形式存在于水溶液中,而溶解在溶液中的Pu则以部分胶体和部分水合无定形Pu(Ⅳ)氧化物形式存在。
5 总结和展望
针对后处理过程中不同的处理目的已经开发出了多个流程和萃取剂,而萃取剂的辐照稳定性很大程度上影响着流程的好坏和处理效果。所以新型萃取剂的合成主要是为了实现更高的萃取效率和更优异的辐射稳定性。由于镧系元素偏软,基于软原子N或S配体的萃取剂成为目前的研究趋势,已经开发出来了二甘醇酰胺(DGAs)、双-三嗪基-吡啶类(BTPs)等含N萃取剂和亚枫等含S萃取剂。一方面通过改性或者接枝到杯冠等结构中以提高萃取性能,另一方面增加保护基团从而使得激发态的萃取剂自由基或离子通过复合、空穴转移或者自由基转移等反应保护关键基团。由于稀释剂的辐解自由基对萃取剂的影响也比较大,人们发现离子液体作为稀释剂时比传统的NDD对萃取剂的影响更小。0.001 mol/L C4DGA/[C8mim][NTf2]型萃取体系的D(Am)高达5 500,在500 kGy的累积辐照后,D(Am)只减少了7%[66]。相比于TBP/NDD等传统萃取体系,该新型萃取体系的萃取性能和辐射稳定性有很大的提高。因此发展新型的离子液体和萃取剂是未来的趋势。
迄今为止,对萃取剂辐射稳定性的研究绝大多数采用稳态辐照后性能表征和稳态产物分析的方法,对硝酸水溶液与镧系元素的水溶液辐解也多采用稳态辐照后对溶液光谱进行分析的方法。然而,对辐解过程中的瞬态产物和短时间尺度内辐解基元反应动力学仍缺乏系统研究,这无疑不利于深入认识其辐解机理。因此,需要加强辐射化学的基础反应过程研究,特别是对短寿命中间产物的研究。飞秒、皮秒级等超快脉冲辐解技术将会有利地促进对新型萃取剂等辐解机理的研究。实际上,许多化学基础反应过程都发生在皮秒甚至亚皮秒时间范围内,如电子、正离子和负离子的溶剂化过程,离子对的复合,液体中激发态的形成和衰减,纳米粒子的成核过程,极度狭窄空间体系中的电子转移和反应等。在乏燃料后处理中广泛使用的烷烃类有机溶剂,介电常数低,Onsager半径大,电离辐射作用后生成的电子很快地复合,因此直接观测的时间在皮秒及亚皮秒范围。被认为在新核能系统、核燃料循环中颇有前景的介质体系如超临界流体、离子液体等,以及在熔盐堆和乏燃料干法后处理中使用的高温熔盐体系等,对这些体系辐解机理的研究,也只有超快脉冲辐解技术才能胜任。
猜你喜欢
今日农业(2020年20期)2020-12-15 15:53:19
核化学与放射化学(2020年4期)2020-08-21 07:45:08
能源(2018年10期)2018-12-08 08:02:48
湿法冶金(2018年2期)2018-04-25 05:08:01
质谱学报(2016年4期)2016-08-02 05:33:25
浙江大学学报(工学版)(2016年9期)2016-06-05 09:20:57
中学生理科应试(2016年2期)2016-05-30 14:23:28
广东石油化工学院学报(2016年3期)2016-05-17 05:16:21
能源(2016年10期)2016-02-28 11:33:30
应用化工(2014年7期)2014-08-09 09:20:28
