
神经酰胺在血管病理生理中作用的研究进展
2020-03-01 07:04张芳妮蒋宏峰艾玎
药学进展 2020年12期
张芳妮,蒋宏峰,艾玎*
(1.天津医科大学生理与病理生理学系,天津 300070;2.北京市心肺血管疾病研究所 首都医科大学附属北京安贞医院,北京 100029)
血管作为血液循环的基础通道和重要场所,连通全身脏器和组织,其正常运行对于保持机体功能的正常运行至关重要。神经酰胺作为鞘脂代谢的核心物质,除了构成细胞质膜结构之外,还承担信号传导的任务,直接或间接影响细胞稳态,与多种疾病的发生和发展关系密切[1]。神经酰胺对血管稳态的调节和血管疾病的发生、发展具有至关重要的作用。本文将对神经酰胺在体内的生成、代谢与分布情况,神经酰胺对血管细胞(包括内皮细胞、平滑肌细胞、成纤维细胞等)的作用及其与高血压、动脉粥样硬化和血管新生等血管疾病的关系进行综述。
1 神经酰胺概述
神经酰胺由鞘氨醇骨架与不同长度的脂肪酸链(以C14~C26 偶数碳链为主)通过酰胺键连接组成[2],属于鞘脂类物质,并且是鞘脂代谢通路的核心分子[3]。因鞘氨醇类骨架和酰基链具有多样性,现已鉴定出数百种神经酰胺[4]。
鞘脂类物质是细胞质膜的重要组成部分,所有人体组织均可产生神经酰胺[5]。神经酰胺在正常细胞质膜中维持非常低的浓度[4],在细胞因子、死亡受体配体和抗癌药物等刺激下,其产生会增加[6]。神经酰胺及其相关的酶也在亚细胞结构中被发现,如Obeid 实验室发现线粒体(非其他细胞器)中的神经酰胺可诱导细胞凋亡[7]。另外,血浆中神经酰胺水平非常低,只占鞘脂含量的3%左右;神经酰胺可与载脂蛋白结合,以极低密度脂蛋白、低密度脂蛋白和高密度脂蛋白的形式分布于血浆中[8]。
神经酰胺生成与代谢相关酶和产物见图1。神经酰胺可由3 条代谢途径生成:1)从头合成途径。此途径是神经酰胺在细胞中的主要来源,所有真核细胞均能够以这种方式产生鞘脂。在内质网胞浆面,丝氨酸棕榈酰转移酶可催化棕榈酰基-辅酶A 和丝氨酸生成3-酮基双氢鞘氨醇,此步为从头合成途径的限速步骤。3-酮基双氢鞘氨醇在还原酶的作用下被还原成二氢鞘氨醇,之后6 种不同的神经酰胺合酶(CerS 1~CerS 6)对二氢鞘氨醇进行N-酰化形成二氢神经酰胺,最后二氢神经酰胺被去饱和酶催化转化为神经酰胺[9]。2)鞘磷脂酶途径。发生在内质网/高尔基复合体、溶酶体及线粒体等多个细胞器中,鞘磷脂可在酸性鞘磷脂酶或中性鞘磷脂酶的催化作用下生成神经酰胺[10]。3)补救途径。此途径也被称为鞘脂循环途径,是多种鞘脂被分解重新形成神经酰胺的过程。此过程发生在溶酶体或内体中,多种复杂鞘脂被酶催化转化为神经酰胺,神经酰胺在酸性神经酰胺酶催化下转化成鞘氨醇,鞘氨醇被回收至内质网,在神经酰胺合酶的催化下重新转化为神经酰胺[11]。
神经酰胺需从内质网转运到高尔基体中进行代谢,现已知有囊泡转运和脂质蛋白转运2 种主要转运机制[12]。若神经酰胺是以转运囊泡的形式从内质网中转运至高尔基体,其可分别被葡糖神经酰胺合酶催化形成葡糖神经酰胺,或被半乳糖神经酰胺合酶催化形成半乳糖神经酰胺;若神经酰胺是以非囊泡的转运蛋白的形式转运至高尔基体,其可被鞘磷脂合酶催化形成鞘磷脂。最近研究发现,在酵母中存在一种Nvj2p 介导的非囊泡转运机制,在内质网应激刺激下,Nvj2p可促进内质网和高尔基体的连接,从而促进神经酰胺转运[13],而人体内Nvj2p 同源蛋白HT008 在应对内质网应激时是否可促进神经酰胺转运还鲜见研究。此外,神经酰胺可在高尔基体中被神经酰胺激酶催化转化为神经酰胺-1-磷酸[8]。在内质网中,神经酰胺可被神经酰胺酶催化形成鞘氨醇,鞘氨醇在鞘氨醇激酶的催化下可生成鞘氨醇-1-磷酸[14]。
既往神经酰胺仅仅被当做细胞结构的成分,现在业界已经认识到神经酰胺及其相关的酶均具有重要生物活性,其既可以参与细胞对外界刺激的反应过程,又可直接调节细胞的生长、分化、衰老及凋亡等。神经酰胺主要通过2 种方式调节病理生理反应:1)作为生物活性分子直接作用。特定的刺激可促进神经酰胺合成,新形成的神经酰胺直接与蛋白质特定结构域作用。2)调节膜物理性质。神经酰胺参与脂筏结构和富含神经酰胺的脂质平台的形成,可改变膜受体构象,最终促进刺激后信号传导的放大[11,15]。目前,神经酰胺在血管相关细胞(如内皮细胞、平滑肌细胞及成纤维细胞等)和血管相关疾病过程(如高血压、动脉粥样硬化等)中的作用已被广泛关注。
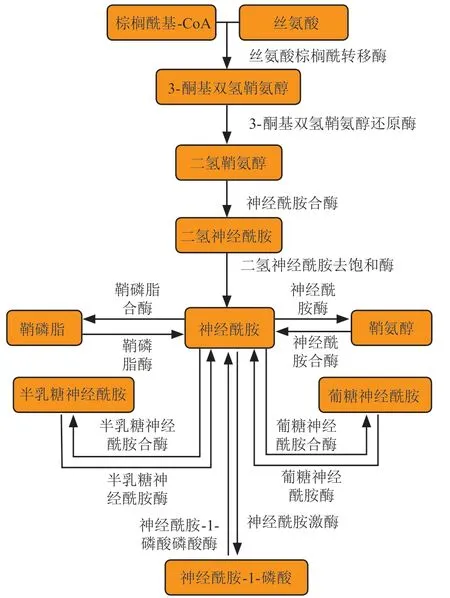
图1 神经酰胺生成与代谢Figure 1 Generation and metabolism of ceramides
2 神经酰胺对血管相关细胞类型的作用
2.1 血管内皮细胞
内皮细胞覆盖整个血管内壁,介导血管通透性、屏障保护和破坏、血管收缩和舒张、应激反应和血管再生等功能。神经酰胺可在内皮细胞中高水平表达,其中人冠状动脉和脐静脉内皮细胞中酸性鞘磷脂酶表达基线水平是巨噬细胞的20 倍,并且在炎症细胞因子白细胞介素-1β(IL-1β)、干扰素-γ(IFN-γ)刺激下鞘磷脂酶分泌量可增加2~3 倍[16]。最新研究表明,内皮细胞从头合成途径产生的神经酰胺是血浆神经酰胺表达水平的关键决定因素,并且血浆神经酰胺水平也可能是内皮细胞是否正常维持血管张力和血压稳态的一个预测因子[17]。
神经酰胺介导内皮细胞凋亡的作用已被证实。研究表明,感染性休克、热休克、电离辐射和氧化应激等均可刺激神经酰胺的产生,从而促进内皮细胞凋亡[18]。神经酰胺可通过激活神经酰胺活化蛋白激酶(CAPK)、Ras 相关因子-1(Raf-1)和核因子кB(NF-кB)等来诱导线粒体细胞色素C 释放或激活半胱天冬酶-8(Caspase-8),也可通过促进促凋亡B 淋巴细胞瘤-2 基因(Bcl-2)家族蛋白的寡聚化来介导内皮细胞凋亡[18]。最新研究表明,神经酰胺通过激活p38 丝裂原活化蛋白激酶(MAPK)通路参与电离辐射诱导的内皮细胞凋亡[19]。另外,C6-神经酰胺可诱导人脐静脉内皮细胞衰老,细胞表现为胞体增大、G1期细胞周期阻滞等[20]。
舒张血管物质如内皮舒张因子—一氧化氮(NO)、前列环素等和收缩血管物质如血管紧张素Ⅱ、内皮素等可被内皮细胞释放以实现血管张力的稳态调节。其中,NO 是影响血管舒缩的最重要物质之一,其在内皮细胞中由内皮型一氧化氮合酶(eNOS)催化L-精氨酸合成[21]。神经酰胺可通过调节内皮细胞中NO 的产生来影响血管张力,改变血管通透性,介导内皮屏障的功能变化,影响血流诱导血管舒张的过程。目前,大部分研究认为神经酰胺可通过抑制内皮细胞中NO 的合成或促进NO 的转化和分解来抑制血管舒张。Trayssac 等[20]研究表明,中性鞘磷脂酶活性随着年龄的增长而增强,可致神经酰胺表达水平升高,最终导致eNOS 失活。神经酰胺促进eNOS 失活的机制可能为:神经酰胺与负性调控因子小窝蛋白-1 结合,还可与胞质中PP2A 抑制剂2(I2PP2A)结合,两者均可促进蛋白激酶B(Akt)-热休克蛋白90(Hsp90)-eNOS 复合物的解离,抑制eNOS 磷酸化和活化[22]。然而,另有研究证实,外源性肿瘤坏死因子-α(TNF-α)急性刺激人内皮细胞可诱导中性鞘磷脂酶的聚集,促进eNOS 的激活[23]。研究显示,在肺动脉内皮细胞中,酸性鞘磷脂酶的激活可促进神经酰胺合成,导致小窝蛋白-1在小窝中积聚,进而抑制eNOS 的活性和NO 的释放,造成血管通透性增强,肺水肿形成[24]。然而,在体循环中,神经酰胺可激活内皮细胞eNOS,促进NO 释放,形成比肺循环更大的细胞间隙,产生间质水肿[18]。此外,在剪切应力诱导血管舒张的过程中,神经酰胺诱导内皮细胞产生的NO 发挥了重要的作用。中性鞘磷脂酶可在肺血流增加时快速激活,使内皮细胞表面神经酰胺表达水平增加,而神经酰胺迅速大量增加可刺激内皮细胞激活Akt/eNOS通路[25]。另有研究表明,神经酰胺慢性刺激小动脉可使介导血流诱导舒张的介质从NO 变为有害的H2O2,而抑制神经酰胺的生成可使NO 产生增多,H2O2产生减少[26]。
神经酰胺可通过触发活性氧(ROS)的形成,增加氧化应激反应来介导内皮细胞功能[23]。研究表明,在Fas 配体和TNF-α 等刺激下,内皮细胞中富含神经酰胺的脂筏发生聚集,从而增强腺嘌呤二核苷酸磷酸(NADPH)氧化酶(Nox)的活性,促进ROS 的产生[27]。
2.2 血管平滑肌细胞
平滑肌细胞在支持血管新生和内膜增生、调节血管张力等方面起着关键作用。神经酰胺可在体外诱导血管平滑肌细胞凋亡已成为共识。研究表明,在NO 诱导血管平滑肌细胞凋亡的过程中,神经酰胺合成增加,酸性鞘磷脂酶抑制剂地昔帕明可抑制NO 诱导的平滑肌细胞凋亡,提示神经酰胺可在平滑肌细胞凋亡过程中充当桥梁的作用[28]。另有研究显示,神经酰胺可通过激活p38MAPK 通路,抑制核蛋白入核来抑制平滑肌细胞增殖[29],但在动脉粥样硬化病变过程中,氧化低密度脂蛋白(OxLDL)可通过基质金属蛋白酶-2(MMP-2)激活鞘磷脂/神经酰胺途径来诱导平滑肌细胞增殖[30]。此外,神经酰胺可通过刺激环氧合酶-2(COX-2)和内质网应激来增强血管平滑肌细胞收缩反应[31]。神经酰胺还可促进血管平滑肌细胞向成骨细胞样表型分化,如:OxLDL 可通过促进人血管平滑肌细胞Toll 样受体4(TLR4)的表达,激活NF-κB/神经酰胺通路,从而促进MSX2、OSTERIX、BMP2和KLF4等成骨分化相关基因的表达[32]。此外,在高磷诱导的平滑肌细胞钙化过程中,相对于正常细胞,过表达酸性鞘磷脂酶的冠脉平滑肌细胞成骨分化相关基因OSP和RUNX2表达增加,细胞内钙沉积增加[33]。
远端肺动脉和动脉导管的血管平滑肌细胞可以感知局部缺氧,促进缺氧性肺血管收缩[34]。研究表明,缺氧条件下中性鞘磷脂酶催化生成的神经酰胺在肺血管平滑肌中迅速增加,其可通过激活蛋白激酶Cζ(PKCζ)来促进NADPH 氧化酶活化,增加ROS 的产生,ROS 可抑制Kv 通道,从而导致膜去极化,激活L 型Ca2+通道和Rho 激酶(RhoK)介导的Ca2+敏化[35]。另外,神经酰胺还可通过囊性纤维化跨膜电导调节器(CFTR)刺激Ca2+,使其通过瞬时受体电位离子通道蛋白6(TRPC6)进入平滑肌细胞内,从而促进血管收缩[36]。
2.3 其他血管相关细胞类型
成纤维细胞是血管外膜中最丰富的细胞,具有很强的增殖和迁移能力,可分泌Ⅰ型和Ⅲ型胶原、纤维连接蛋白等细胞外基质,在血管重塑过程中起着重要作用[37]。最新研究表明,血管外膜成纤维细胞可表达酸性鞘磷脂酶,沉默酸性鞘磷脂酶基因可减少脂筏中神经酰胺的聚集,抑制血管紧张素Ⅱ刺激的血管外膜成纤维细胞增殖细胞核抗原(PCNA)表达,降低Ⅰ型胶原水平,显著减少细胞迁移,提示酸性鞘磷脂酶和神经酰胺可介导血管外膜重塑中成纤维细胞的增殖、迁移和细胞外基质成分的分泌[38]。另外,血管相关巨噬细胞亦受到神经酰胺的调节,神经酰胺可减少巨噬细胞CD36 的表达,从而减少OxLDL 的摄取[39]。Singh 等[40]发现,在形成泡沫细胞的过程中,巨噬细胞可积聚神经酰胺并通过激活RhoA/RhoK 信号通路来抑制肌动蛋白聚合和泡沫细胞形成。
3 神经酰胺与血管相关疾病的关系
3.1 神经酰胺与高血压
高血压由血管异常收缩/舒张、血管重塑和动脉血压升高所致。大多数研究认为,血浆神经酰胺水平与高血压的严重程度呈正相关[41]。Spijkers 等[42]研究发现,原发性高血压患者血浆神经酰胺水平高于正常人;与血压正常大鼠相比,自发性高血压大鼠(Spontaneously Hypertensive Rat)动脉组织和血浆中神经酰胺含量均升高。丝氨酸棕榈酰转移酶抑制剂myriocin 的使用或二氢神经酰胺去饱和酶的杂合性缺失均可保护小鼠免受高脂诱导的血管功能损伤,从而改善高血压[43]。最新研究发现,内皮细胞特异性丝氨酸棕榈酰转移酶长链亚基2(Sptlc2)敲除小鼠中,从头合成途径神经酰胺的产生被抑制,从而导致小鼠肠系膜动脉eNOS 活性增强,最终促使血压下降[17]。除了影响NO 的释放外,神经酰胺可通过分子间氢键增加细胞膜的硬度,从而降低细胞膜的流动性来促进血管收缩[44]。
3.2 神经酰胺与动脉粥样硬化
已有研究证实,动脉粥样硬化患者血浆中神经酰胺水平升高,在粥样硬化斑块区域可分离出包含神经酰胺在内的多种鞘脂,且斑块LDL 中神经酰胺的含量比在血浆LDL 中高10~50 倍[40,45]。最新研究表明,血浆神经酰胺水平越高的急性心肌梗死患者斑块易损特征越明显[46]。一些前瞻性研究已证实,血浆中Cer(d18:1/16:0)、Cer(d18:1/20:0)、Cer(d18:1/24:1)及其与Cer(d18:1/24:0)的比率对斑块不稳定性、冠心病病死率和健康人发生意外主要心血管不良事件(MACE)均具有预测能力[47-50]。Myriocin 可降低ApoE-/-小鼠血脂水平,阻止粥样硬化病变的进展,提示抑制催化生成神经酰胺的酶可显著减少动脉粥样硬化病变[8]。
神经酰胺可通过调控炎症反应来促进动脉粥样硬化的发生。Edsfeldt 等[51]证实,人颈动脉斑块中神经酰胺的表达水平与白细胞介素-6(IL-6)和单核细胞趋化蛋白-1(MCP-1)的表达水平强相关,神经酰胺体外刺激人冠状动脉平滑肌细胞可促进IL-6的产生。Koka 等[52]证实,酸性鞘磷脂酶和神经酰胺信号通路可促进炎症小体NLRP3 的激活,从而导致动脉粥样硬化的发生。此外,在动脉中,神经酰胺可促进脂蛋白进入血管壁[5],然而Singh 等[39-40]发现,斑块中巨噬细胞可积聚神经酰胺,神经酰胺可减少肌动蛋白的聚合,减少LDL 的吞噬,从而抑制泡沫细胞生成,此外被吞噬的LDL 中富含神经酰胺,神经酰胺含量的增高更加促使泡沫细胞的生成减少。
3.3 神经酰胺与血管新生
血管新生是在原有的微循环的基础上形成新的血管,其参与生理发育、伤口愈合和肿瘤发生等多个病理生理过程,主要特征为内皮细胞的增殖、迁移、分化和成熟[53]。在血管新生过程中,血管内皮生长因子(VEGF)等血管新生诱导因子和血管抑素、内皮抑素、基质金属蛋白酶等抑制因子相互制约。其中,VEGF 是研究最广泛的血管新生诱导因子[54]。研究显示,大麻素通过介导神经酰胺/p8 途径可抑制VEGF 形成和VEGF 受体2 激活,从而影响肿瘤血管新生[55]。C6-神经酰胺可介导低浓度OxLDL 诱导的血管新生[56]。目前,有关神经酰胺影响血管新生的研究较少,大多数研究着重于探究与神经酰胺相关的鞘脂酶类对血管新生的影响,如:酸性神经酰胺酶可通过调控Akt 和细胞外调节蛋白激酶1/2(ERK 1/2)途径抑制血管生成[57]。Camare 等[56]发现,低浓度OxLDL 可促进ROS 诱导的人微血管内皮细胞中的中性鞘磷脂酶-2/鞘氨醇激酶-1 信号通路激活,促进血管新生。
3.4 神经酰胺与血管重塑
最新研究表明,失重大鼠大脑动脉中平滑肌细胞凋亡减少,增殖增加,内膜中层厚度和中膜截面积增加,酸性鞘磷脂酶表达减少,神经酰胺水平降低,肠系膜小动脉中以上相关指标变化情况与之相反,而神经酰胺孵育失重大鼠大脑动脉可以逆转平滑肌细胞凋亡和增殖的变化,提示神经酰胺对平滑肌细胞凋亡和增殖的影响介导大鼠在失重条件下的血管重塑[58]。血管内膜增生属于病理性血管重塑的一种特殊类型,与血管机械损伤、高剪切应力等因素有关,常见于静脉移植、血管成形术和动静脉瘘形成等。血管平滑肌细胞的分化、增殖和迁移在血管内膜增生过程中起重要作用[59-60]。研究显示,神经酰胺涂层的球囊导管显著减少了兔颈动脉在球囊成形术后的内膜增生,这种抑制作用是通过神经酰胺渗入动脉内膜和中膜,减少了处于细胞增殖周期的血管平滑肌细胞的数量而实现的[61]。另外,沉默血管外膜成纤维细胞酸性鞘磷脂酶可减少脂筏中神经酰胺聚集,阻断Nox2 依赖性超氧化物的产生,从而抑制血管外膜重塑[38]。
3.5 神经酰胺与血管钙化
血管钙化是一种钙盐以羟磷灰石的形式在血管壁的沉积结果,是导致动脉粥样硬化、高血压等疾病的重要诱因,主要病理过程包含基质小泡(来源于血管平滑肌细胞衍生的凋亡小体,位于胞外,含有羟磷灰石颗粒,与胞外基质蛋白相互作用启动钙化)形成、细胞外基质钙盐沉积、血管平滑肌细胞凋亡和向成骨细胞分化等[62]。OxLDL 可促进人血管平滑肌细胞成骨分化相关基因表达、早期成骨分化标志物碱性磷酸酶(ALP)活性增加和细胞内钙沉积增多,这些病理过程均有神经酰胺的参与[32]。此外,神经酰胺还可激活p38MAPK 通路,刺激平滑肌细胞凋亡,从而促进血管钙化[63]。平滑肌细胞特异性过表达酸性鞘磷脂酶的小鼠动脉内壁钙化增强,平滑肌细胞骨相关蛋白表达增加,基质小泡分泌增加[33]。
3.6 神经酰胺与其他血管相关疾病过程
2019 年发表的一项研究结果表明,神经酰胺在不同分型急性主动脉夹层患者血清中表达水平存在差异。与健康对照组相比,Stanford A 型患者神经酰胺表达水平降低,而Stanford B 型患者表达差异无统计学意义,提示神经酰胺可能是区分不同Stanford分型急性主动脉夹层患者的潜在标志物[64]。Meher 等[65]发现,神经酰胺介导IL-1β 诱导的中性粒细胞胞外陷阱的形成,可促进小鼠腹主动脉瘤的发生。另外,酸性鞘磷脂酶作为神经酰胺的重要产生来源,在川崎病患者血清中水平显著升高,提示包含神经酰胺在内的鞘脂通路是影响川崎病的重要因素[66]。
4 总结和展望
综上所述,高血压、动脉粥样硬化等血管病变患者血浆神经酰胺显著升高,神经酰胺有望成为冠心病等疾病的风险预测因子;动脉夹层、腹主动脉瘤和血管炎的形成与神经酰胺有关;神经酰胺介导内皮细胞的衰老及凋亡,参与氧化应激、NO 的合成和释放等过程,进而调控血管收缩舒张、血管通透和血管新生等。血管平滑肌细胞的增殖、凋亡、表型转化、缺氧感知等均受到神经酰胺的调控,影响血管收缩舒张、血管钙化、血管斑块形成与病变以及血管重塑等;血管外膜重塑中成纤维细胞的增殖和迁移也受神经酰胺的调控;中性粒细胞胞外陷阱和巨噬细胞形成泡沫细胞的过程均受到神经酰胺的调节;与神经酰胺生成和分解相关的酶如中性鞘磷脂酶、酸性鞘磷脂酶和丝氨酸棕榈酰转移酶等常被用来作为预测神经酰胺表达水平和调控神经酰胺在体内表达水平变化的切入点和靶点。大多数研究认为,神经酰胺对血管发挥有害作用,但也有少部分研究表明其有益,因此,神经酰胺在机体中调控血管作用的机制错综复杂,尚无定论。此外,作为鞘脂代谢过程的重要成员,神经酰胺相关酶类的活性与神经酰胺在机体内的变化息息相关,神经酰胺及其相关酶类可为探索血管相关疾病标志物和寻找相应治疗靶点提供参考。
猜你喜欢
医学综述(2021年24期)2022-01-11
现代临床医学(2021年5期)2021-11-02
中国心血管杂志(2020年2期)2020-05-15
安徽医科大学学报(2020年1期)2020-02-14
中国眼镜科技杂志(2019年9期)2019-11-11
中国临床医学影像杂志(2019年4期)2019-06-18
浙江中西医结合杂志(2018年12期)2018-12-27
天然产物研究与开发(2018年11期)2018-11-30
中国麻风皮肤病杂志(2017年10期)2017-11-06
中国病理生理杂志(2015年8期)2015-12-21
