
神经酰胺代谢紊乱在非酒精性脂肪性肝病发生发展中的作用
2022-01-11 09:42李亚平赵格郭子仪杨文琦
医学综述 2021年24期
李亚平,赵格,郭子仪,杨文琦
(广州体育学院,广州 510000)
非酒精性脂肪性肝病(non-alcoholic fatty liver disease,NAFLD)是指由酒精摄入以外的因素(如高脂饮食、久坐)所致的慢性肝脏疾病,脂质在肝细胞内过量蓄积是该病的重要标志[1]。NALFD由最初的单纯性脂肪变性可发展为非酒精性脂肪性肝炎、肝纤维化、肝硬化甚至肝细胞癌[2]。NAFLD作为一种代谢综合征,其发病机制复杂。James于1998年提出的“二次打击”学说是NAFLD发病机制的经典假说[3]。“第一次打击”主要是指肥胖、胰岛素抵抗等病理因素所致的肝细胞内脂质蓄积,随后由脂质蓄积诱导氧化应激进而引起肝脏炎症,即为“第二次打击”[3]。神经酰胺是由不同链长的脂肪酸和鞘氨醇通过酰胺键连接形成的一类脂质,在细胞凋亡、增殖、分化等生理过程中发挥重要作用。神经酰胺代谢异常与NAFLD的发生、发展密切相关,不同种类神经酰胺发挥不同作用。现就神经酰胺代谢在NAFLD发生发展中的作用进行综述。
1 神经酰胺的代谢
神经酰胺由鞘氨醇与不同链长脂肪酸通过酰胺键构成,是鞘脂代谢的中间产物和构成生物膜的重要脂质。神经酰胺主要有3条合成途径:从头合成途径、神经鞘磷脂分解代谢途径、补救途径[4],见图1。
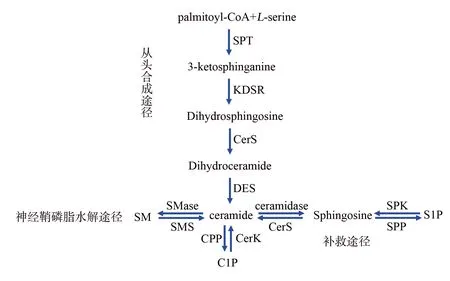
palmitoyl-CoA:棕榈酸辅酶A;L-serine:L-丝氨酸;SPT:丝氨酸棕榈酰转移酶;3-ketosphinganine:3-酮基二氢鞘氨醇;KDSR:酮基二氢鞘氨醇还原酶;Dihydrosphingosine:二氢鞘氨醇;CerS:神经酰胺合成酶;Dihydroceramide:二氢神经酰胺;DES:二氢神经酰胺脱氢酶;SM:神经鞘磷脂;SMase:神经鞘磷脂酶;SMS:神经鞘磷脂合成酶;ceramide:神经酰胺;ceramidase:神经酰胺酶;Sphingosine:鞘氨醇;SPK:鞘氨醇激酶;SPP:鞘氨醇-1-磷酸磷酸酶;S1P:鞘氨醇-1-磷酸;CPP:神经酰胺-1-磷酸磷酸酶;CerK:神经酰胺激酶;C1P:神经酰胺-1-磷酸
1.1神经酰胺从头合成途径 该途径主要发生在内质网。首先棕榈酰辅酶A与L-丝氨酸在丝氨酸棕榈酰转移酶(serine palmitoyltransferase,SPT)催化下生成不稳定分子3-酮基二氢鞘氨醇[4]。随后3-酮基二氢鞘氨醇在3-酮基二氢鞘氨醇还原酶催化下生成二氢鞘氨醇,二氢鞘氨醇在神经酰胺合成酶(ceramide synthase,CerS)的催化下N酰基化生成二氢神经酰胺。二氢神经酰胺在二氢神经酰胺脱氢酶(dihydroceramide desaturase,DES)的催化下生成神经酰胺[5]。神经酰胺一旦生成,即通过囊泡或神经酰胺转移蛋白从内质网转移至高尔基体转化为神经鞘磷脂[1,5]。CerS是神经酰胺从头合成的关键酶,主要位于内质网。在哺乳动物中发现有6个亚型(CerS1~CerS6),每一个亚型控制不同脂肪链长度的神经酰胺合成。CerS1主要控制C18神经酰胺合成,CerS2主要控制C22~C24神经酰胺合成,CerS3主要控制超长链神经酰胺C26~C34的合成,CerS4主要控制C18~C20神经酰胺合成,CerS5和CerS6主要控制C14~C16神经酰胺的合成[6]。
1.2神经鞘磷脂分解代谢途径 神经鞘磷脂与神经酰胺可分别在神经鞘磷脂酶及神经鞘磷脂合成酶作用下相互转化[7]。神经鞘磷脂酶主要有中性神经鞘磷脂酶、酸性神经鞘磷脂酶和碱性神经鞘磷脂酶,其中酸性神经鞘磷脂酶对NAFLD的发生发展起重要作用。
1.3补救通路 该通路主要发生在溶酶体,是指某些复杂鞘脂(如鞘氨醇-1-磷酸)在鞘氨醇-1-磷酸磷酸酶作用下生成鞘氨醇,随后鞘氨醇在CerS的作用下生成神经酰胺[7]。神经酰胺与复杂鞘脂间可互相转化。神经酰胺可在神经酰胺酶作用下生成鞘氨醇,而后在鞘氨醇-1-磷酸激酶的催化下合成鞘氨醇-1-磷酸盐。
2 神经酰胺代谢与NAFLD的关系
神经酰胺代谢紊乱贯穿NAFLD的各个阶段[8]。NAFLD患者[9]及动物模型[10]血液和肝脏中总神经酰胺含量增加。采用基因敲除或药物抑制神经酰胺从头合成限速酶SPT的表达,可降低肝脏与血液总神经酰胺水平并改善NAFLD[11]。随着NAFLD的发展,肝脏不同种类神经酰胺存在不同变化,其中C16长链神经酰胺水平逐渐增加,C22~C24超长链神经酰胺逐渐减少[11-12]。国外研究表明,敲除CerS6[13]、CerS5[14]可通过降低C16神经酰胺含量抑制NAFLD的发展,提示C16神经酰胺可能促进NAFLD的发生发展。Kim等[12]通过细胞转染技术使小鼠肝细胞CerS2过表达,发现超长链神经酰胺含量显著升高,CerS6和长链神经酰胺则与之相反,且内质网应激和胰岛素抵抗得到改善。提示超长链神经酰胺含量增加可能通过抑制长链神经酰胺的合成而发挥有益作用。因此,肝脏长链神经酰胺与超长链神经酰胺的比例可能在NAFLD的发生发展中发挥关键作用。
3 神经酰胺在NAFLD病理机制中的作用
胰岛素抵抗、内质网应激、线粒体氧化应激、肝细胞凋亡、炎症反应、肝纤维化均已被证实是NAFLD发生发展过程中的重要病理机制。肝细胞脂质蓄积,尤其是神经酰胺的蓄积与NAFLD的发生发展密切相关。
3.1神经酰胺与胰岛素抵抗 胰岛素抵抗是NAFLD发生发展的主要机制。肝细胞内脂质(如神经酰胺、二酰甘油、三酰甘油)蓄积是导致肝胰岛素抵抗的主要因素[15],其中神经酰胺是学术界公认的可调控胰岛素抵抗的脂质。采用多球壳菌素抑制神经酰胺从头合成途径可改善肝脏胰岛素抵抗,缓解肝损伤[16]。SPT和DES是神经酰胺从头合成途径的关键酶,SPT长链碱性亚基3和DES-1分别是控制SPT和DES表达的基因。SPT长链碱性亚基3和DES-1基因敲除实验表明,抑制神经酰胺从头合成可增加肝脏胰岛素敏感性,改善NAFLD脂肪变性[17-19]。目前神经酰胺调控胰岛素抵抗的机制尚不明确。有研究表明,神经酰胺可能通过抑制丝氨酸/苏氨酸激酶通路,抑制胰岛素功能,从而促进胰岛素抵抗[20-22]。神经酰胺抑制丝氨酸/苏氨酸激酶通路的可能机制包括:激活脑内富集的Ras同系物/哺乳动物雷帕霉素靶蛋白复合物1/S6激酶信号抑制丝氨酸/苏氨酸激酶磷酸化[21];激活蛋白磷酸酶2A刺激丝氨酸/苏氨酸激酶脱磷酸化[23];促进蛋白激酶Cζ磷酸化破坏丝氨酸/苏氨酸激酶的转移[24]。
胰岛素抵抗可进一步加重脂代谢紊乱[25]。已有研究证实,胰岛素抵抗会激活碳水化合物反应元件结合蛋白和甾醇调节元件结合蛋白1c,进而促进肝脏脂质蓄积[26]。神经酰胺作为体内重要的一种脂质,其代谢也可能受胰岛素相关通路的调控,胰岛素抵抗是否会促进神经酰胺代谢紊乱仍不清楚。
3.2神经酰胺与内质网应激 内质网应激在NAFLD的发生发展中起重要作用。脂质代谢异常尤其是神经酰胺代谢紊乱是诱发内质网应激的重要机制。NAFLD常伴随着肝脏总神经酰胺增加及内质网应激加重[27],其中不同神经酰胺的变化有所不同:C16神经酰胺水平有所增加,C22~C24神经酰胺含量则降低[12]。动物实验[27]及人类细胞实验[12,27]均表明,降低肝脏总神经酰胺含量可减轻内质网应激程度和NAFLD。近年来的研究提示,不同酰基链长度的神经酰胺可能对内质网应激有不同的调节作用,如C16、C18神经酰胺可诱导内质网应激,加重NAFLD;而C22、C24神经酰胺则与之相反[12]。
内质网是蛋白质合成和脂质代谢的场所,肝脏持续的内质网应激除导致肝细胞凋亡、炎症、胰岛素抵抗等病理特征外,还会破坏内质网调节脂质代谢的能力,进而加重脂代谢紊乱,促进NAFLD的发展。但内质网应激是否通过加重神经酰胺代谢紊乱促进NAFLD的发展尚不明确,有待进一步研究。
神经酰胺调控内质网应激的机制尚不完全清楚,可能与神经酰胺改变内质网钙离子稳态有关[28]。有研究表明,激活AMP活化的蛋白激酶可以改善内质网应激[29],星形胶质细胞内短链C2神经酰胺可增加AMP活化的蛋白激酶表达[30]。但神经酰胺能否通过调控AMP的活化蛋白激酶通路而调节内质网应激仍不清楚,需进一步研究。
3.3神经酰胺与线粒体氧化应激 氧化应激是指活性氧类过量产生所引起的组织损伤。线粒体氧化应激是NAFLD的重要病理机制[31]。线粒体是活性氧类产生及脂肪酸氧化的重要场所。肝线粒体氧化应激可导致线粒体结构及功能受损,从而引发神经酰胺、三酰甘油、二酰甘油等脂质在肝细胞蓄积。肝细胞脂质(神经酰胺[32])蓄积又会导致活性氧类的过量产生,进一步加重线粒体氧化应激[33]。尽管尚缺乏总神经酰胺含量增加加重NAFLD肝细胞线粒体氧化应激的直接证据,但已在足细胞、卵母细胞、白细胞及骨骼肌中证实总神经酰胺含量增加可加剧线粒体氧化应激[34-35]。C16神经酰胺增加可能促进肝线粒体氧化应激[36]。神经酰胺总量以及不同种类神经酰胺(如超长链神经酰胺)对NAFLD线粒体氧化应激的影响有待进一步研究。
神经酰胺诱导线粒体氧化应激机制尚不明确,已有的研究表明,神经酰胺可直接抑制线粒体呼吸链复合物4的活性,增加活性氧类,导致线粒体氧化应激[36];神经酰胺可导致线粒体呼吸链过氧化氢增加[37]或激活Ras相关C3肉毒毒素底物1(Ras-related C3 botulinum toxin substrate 1,rac1)[38]诱导线粒体氧化应激。
3.4神经酰胺与肝细胞凋亡 肝细胞凋亡是NAFLD的主要特征之一[39]。神经酰胺作为第二信使在肝细胞凋亡过程中发挥重要作用[40]。NAFLD大鼠肝脏神经酰胺总量升高伴随着肝细胞凋亡增多[41],其中主要是C16神经酰胺含量增加[42]。小鼠多球壳菌素干预实验提示,降低肝总神经酰胺水平可减少肝细胞凋亡并改善NAFLD[41]。神经酰胺调控肝细胞凋亡的可能机制包括:①神经酰胺可能通过上调促凋亡因子Bax和Bad,下调抗凋亡因子Bcl-2和Bcl-xL,破坏线粒体膜通透性并释放细胞色素C,进而导致肝细胞凋亡[43]。②神经酰胺可通过激活p53上调的凋亡调控因子导致细胞凋亡[44],p53上调的凋亡调控因子是Bcl-2家族蛋白中唯BH-3域蛋白的成员,其在促进细胞凋亡中发挥重要作用[45]。③神经酰胺通过激活c-Jun氨基端激酶通路上的c-Jun、激活转录因子-2和CCAAT/增强子结合蛋白同源蛋白/生长停滞和DNA损伤诱导基因153而诱导细胞凋亡[40]。
3.5神经酰胺与炎症反应 炎症是一种身体调节反应,过度炎症会加重肝细胞损伤,引起非酒精性脂肪性肝炎并向肝纤维化及肝硬化发展[46-47]。神经酰胺可通过调控肝脏炎症反应而影响NAFLD的发展。研究表明,利用多球壳菌素或脱甲丙咪嗪减少从头合成途径与神经鞘磷脂分解代谢途径产生的神经酰胺,可减轻肝脏炎症反应并改善肝损伤[41,48-49]。近年来的研究提示,神经酰胺可调控肝细胞炎症因子的表达和分泌,且C16神经酰胺是诱导肝脏炎症的主要神经酰胺[49]。在肝细胞中CerS1/2/4/5/6过表达,仅CerS6过表达引起C16神经酰胺含量增加时,肝细胞释放的炎症因子肿瘤坏死因子-α明显上升[49]。提示CerS6可能是C16神经酰胺促进炎症反应的关键蛋白。除普通肝细胞外,肝巨噬细胞对炎症反应的发生也具有重要作用。其中肝巨噬库普弗细胞在调控肝脏炎症反应中发挥重要作用。研究表明,提高库普弗细胞内源性C16神经酰胺的生成可显著增强库普弗细胞吞噬功能[50],提示神经酰胺可能通过作用于库普弗细胞影响肝脏炎症反应。根据活化状态和发挥功能不同可将库普弗细胞与普通巨噬细胞分为M1型和M2型。M1型可分泌多种促炎因子发挥促炎作用,而M2型则可分泌抗炎因子抑制炎症的发生发展,M1与M2表型在一定条件可以互相转换,从而调控炎症反应[51]。神经酰胺能够促进大肠M2型巨噬细胞向M1型巨噬细胞转变,从而加剧炎症反应[52],但神经酰胺对于库普弗细胞表型及炎症因子分泌的影响有待进一步研究。
3.6神经酰胺与肝纤维化 肝纤维化是指非活化肝星状细胞被激活所引起的一种慢性伤口愈合反应。若肝纤维化不能被阻止,最终会发展为肝硬化。抑制肝星状细胞活化或促进活化肝星状细胞凋亡可改善肝纤维化。神经酰胺可调控肝星状细胞的活性及凋亡,但对肝纤维化的作用仍存在争议:①动物实验表明通过药物干预抑制神经酰胺合成的关键酶,可降低肝总神经酰胺水平并改善NAFLD肝纤维化[41]。②动物及细胞实验对肝星状细胞作基因敲除或药物干预均表明,肝星状细胞总神经酰胺积累可改善早期非酒精性脂肪性肝炎肝纤维化[53-54]。这两种相反的结果可能与神经酰胺通过不同通路调控肝星状细胞凋亡及活性有关。在第一种情况中,NAFLD小鼠总神经酰胺增加并不主要促进肝星状细胞凋亡,反而是通过核因子κB[55]炎症反应通路升高促纤维化因子α肌动蛋白-2和α1型胶原蛋白的表达,从而增加肝星状细胞活化数量加剧肝纤维化,这解释了肝总神经酰胺水平增加可加剧肝纤维化这一观点。相反,第二种情况下,小鼠体内特异性促进肝星状细胞神经酰胺合成以及体外培养肝星状细胞增加总神经酰胺含量,可能主要是通过促进肝星状细胞凋亡改善肝纤维化,这解释了肝总神经酰胺增加可改善肝纤维化这一观点。这两种观点均证明抑制肝星状细胞活化或促进活化肝星状细胞凋亡可作为改善NAFLD肝纤维化的靶点,但神经酰胺对肝星状细胞的作用及机制仍需要深入研究。
4 小 结
NAFLD发病的重要细胞机制(胰岛素抵抗、内质网应激、线粒体氧化应激、细胞凋亡、炎症反应和纤维化)均可影响神经酰胺代谢。不同神经酰胺在NAFLD发生发展中的作用并不一致,其中长链神经酰胺(如C16和C18)与超长链神经酰胺(如C22和C24)的比例可能是NAFLD发生的关键因素。因此,调控神经酰胺代谢有望成为治疗NAFLD的有效靶点,但存在以下难点:①SPT是机体的重要代谢酶,利用药物抑制其表达可降低总神经酰胺水平,但由于降低了重要神经酰胺水平,机体正常功能可能受到影响。②特异性抑制肝脏CerS2(调控超长链神经酰胺合成的关键酶)表达可能会引起长链神经酰胺的变化,而其变化对机体产生何种效应仍不明确。因此,高效调控神经酰胺代谢(恢复长链与超长链神经酰胺的正常比例)进而改善NAFLD可能是未来的研究重点。
猜你喜欢
医学研究生学报(2022年3期)2022-11-27
临床肺科杂志(2022年3期)2022-11-26
中华实用诊断与治疗杂志(2022年1期)2022-08-31
传染病信息(2022年2期)2022-07-15
中国典型病例大全(2022年13期)2022-05-10
昆明医科大学学报(2022年2期)2022-03-29
昆明医科大学学报(2022年1期)2022-02-28
医学研究杂志(2021年12期)2021-11-30
中国卒中杂志(2021年7期)2021-11-29
中国体育科技(2018年6期)2018-12-13
