
脑组织生理药代动力学模型研究进展
2020-03-01 07:04桑澜徐胜何华柳晓泉
药学进展 2020年12期
桑澜,徐胜,何华,柳晓泉
(中国药科大学药物代谢动力学研究中心,江苏 南京 211198)
对于作用靶点在中枢神经系统(central nervous system,CNS)的药物而言,脑内浓度直接影响药效;而对于非脑组织靶向的药物,过多的脑内分布可能会引起CNS 不良反应的发生。因此,了解药物在脑内的转运与分布情况在新药研发中十分重要。人脑内的药物浓度难以通过现有分析技术直接测定,而根据脑组织的生理结构和药物特性用生理药代动力学(physiologically-based pharmacokinetic,PBPK)模型来模拟药物在脑内的转运与分布情况是一个可行的策略。本文综述了影响药物在脑内转运分布的生理因素,以及近年来文献报道的研究药物脑组织分布的PBPK 模型,并探讨这类PBPK 模型在药物研发中的应用。
1 影响药物在脑内转运与分布的生理因素
药物在脑内的分布情况受到大脑特殊结构的影响(见图1)。脑皮层下的毛细血管丰富而密集,为血液与脑进行物质交换提供生理基础,进入脑实质后,药物的分布依赖于脑内液体,即脑细胞外液(extracellular fluid,ECF)与脑脊液(cerebrospinal fluid,CSF)的流动;而脑内特有的血-脑屏障(blood-brain barrier,BBB) 和血-脑脊液屏障(blood-cerebrospinal fluid barrier,BCSFB)限制了有害物质进入脑组织,为脑提供保护作用;ECF 和脑细胞统称为脑实质,脑实质被CSF 包围着。药物通过动脉输送到脑部,随后进入毛细血管;屏障上紧密排布的细胞和多种摄取或外排转运体则限制了药物的脑内分布;此外,药物在脑内可能与靶点结合而不能进一步分布。因此,脑内微循环、屏障系统以及药物与靶点的结合是影响药物脑内分布的重要因素。
1.1 脑内微循环
脑内微循环由脑血流、ECF 与CSF 构成。在大脑表面存在大量的动脉和静脉,它们负责输送氧气和营养物质进入脑组织,并将排出的有害物质带离脑组织。部分由大动脉分支后形成的小动脉能够穿透大脑皮层进入毛细血管床。脑部的毛细血管表面积大且非常密集,是血液与脑组织进行物质交换的主要场所。此外,为了维持大脑内的稳态,定期清除脑内有害物质,ECF 与CSF 在脑内不断循环[1]。而药物随着脑血流、ECF 与CSF 的流动完成转运,因此脑微循环内液体流动的方向即为药物转运的方向。
脑微循环中的药物首先通过大血管中的脑血流输送,然后通过脑毛细血管血流流向大脑。因此,血流速度(Qbrain)对于药物向大脑的传递十分重要。在大动脉和静脉中,血流速度约为750 mL·min-1。然而,在与脑组织交换药物的脑毛细血管内,毛细血管血流速度仅为6~12 nL·min-1[2-3];在脉络丛部位,血流速度约为8 mL·min-1[4]。
ECF 约占脑实质的20%,成分与血浆相似,但蛋白质含量较低。BBB 两侧存在离子浓度差时,毛细血管内的血液会流向脑实质,由于蛋白质不能通过BBB,血浆在透过BBB 时被内皮细胞壁过滤形成ECF。在压力差的驱动下,脑ECF 通过对流(ECF bulk flow)在细胞外空间内流动。脑ECF 可以直接流向脑室和蛛网膜下腔(subarachnoid space,SAS)并进入CSF,也可以穿透毛细血管和动脉壁流入淋巴系统。
CSF 是一种蛋白质浓度较低、成分与ECF 相似的透明液体,它由脉络丛上皮细胞分泌产生,脉络丛排列在2 个侧脑室(lateral ventricle,LV)和第3 脑室(third ventricle,TV)、第4 脑室(forth ventricle,FV)的空腔中。依次流经4 个脑室后,CSF 进入小脑延髓池(cisterna magna,CM)并向下流入脊髓;此外,CSF 也可以在 SAS 向上流过大脑表面,通过蛛网膜绒毛中的瓣膜被吸收到静脉中,完成脑脊液循环。
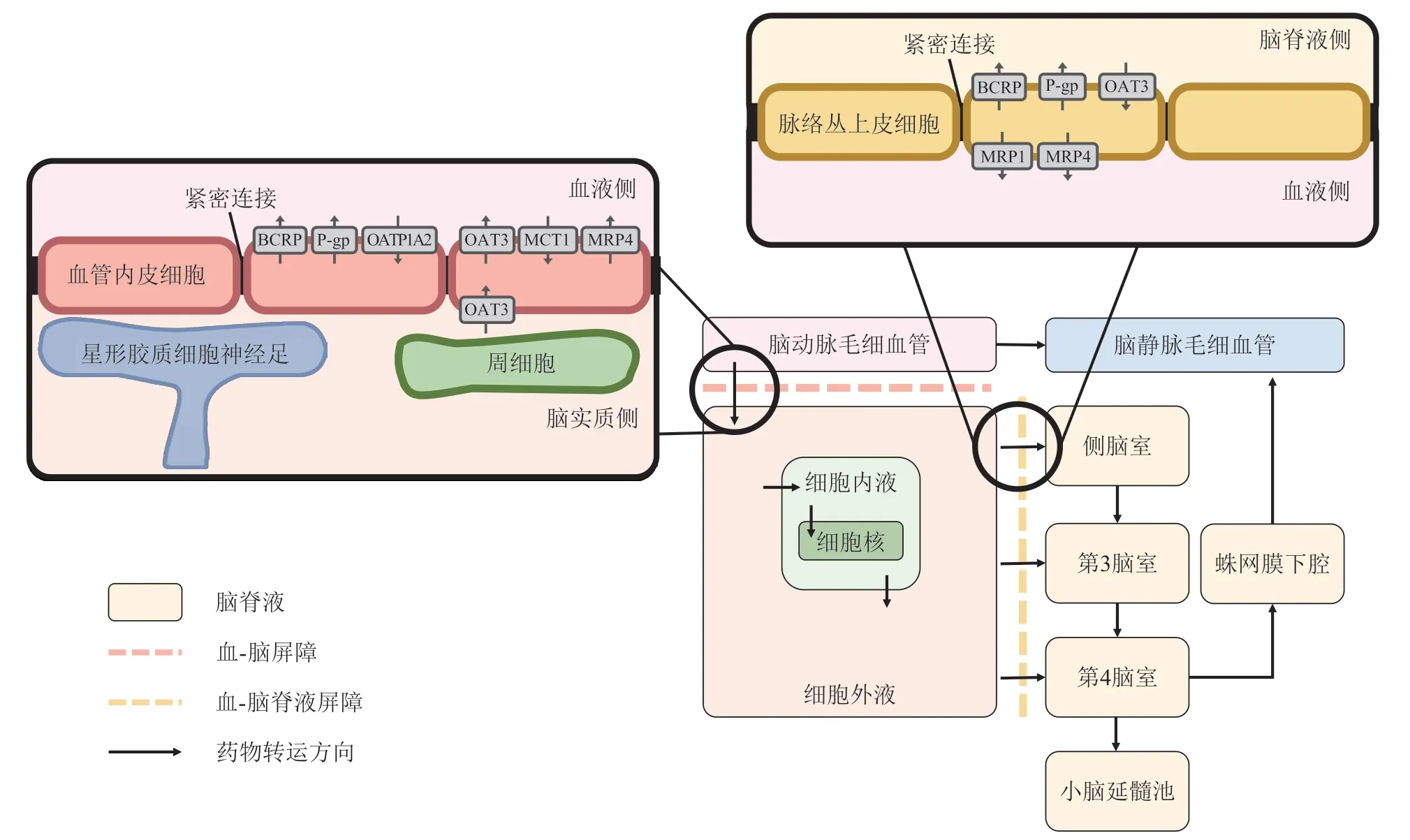
图1 脑的特殊结构及药物在脑内的转运示意图Figure 1 Schematic diagram of brain tissue and drug transport within the brain
1.2 屏障系统与屏障上的药物转运
脑毛细血管血液中的药物在进入脑组织时受到屏障系统的阻碍。在目前的脑组织PBPK 模型中主要考虑BBB 与BCSFB 的作用。BBB 的功能是将血液与脑实质分隔开来,从而避免有害物质进入脑内造成损伤。BBB 上的紧密连接结构、细胞间的多种转运蛋白和缺少窗孔的特征都使得药物难以通过细胞旁途径穿过血管内皮进入脑组织。BBB 功能会随机体的生理病理情况而变化。在某些疾病条件下(比如胶质母细胞瘤),BBB 的紧密连接被破坏,大脑内皮细胞之间的间隙变大,这使得细胞旁转运增加,特别是那些通常不能透过BBB 的大分子[5]。在这类恶性肿瘤的药物开发中,往往不考虑治疗药物的BBB 通透能力,而认为它们对脑组织具有对其他组织同样的渗透性。然而,大量临床证据表明,胶质母细胞瘤患者的BBB 的破坏是不均一的[6]。这使得抗肿瘤药物在BBB 完整的区域达不到理想药效,因此有研究者提出通过聚焦超声等物理手段破坏BBB可以提高BBB 的通透性,增加药物向脑部的转运,实现抗脑肿瘤药物的靶向治疗[7-8]。BCSFB 将脑毛细血管中的血液与脑脊液分离。BCSFB 由脑室的脉络丛上皮细胞紧密排布形成,屏障上的紧密连接阻止了水溶性分子通过细胞旁途径透过BCSFB。与BBB 不同,BCSFB 具有开孔结构和较高的通透性。有研究表明,阿尔茨海默病患者BCSFB 的通透性与转运体活性受到影响,而这可能与脑内炎症因子的沉积有关[9]。
药物在屏障上的转运可以分为被动扩散和主动转运两大类,其中被动扩散包括跨细胞和细胞旁途径,而主动转运主要指通过转运体进行的物质转运。被动扩散是由药物在血液和ECF 中的浓度梯度驱动的,速率取决于屏障两侧的浓度差。药物穿过屏障的难易程度取决于屏障对药物的渗透性(permeability,P)。这种渗透性由屏障自身的通透性(构成屏障的细胞间隙)和药物特性(分子大小和脂溶性)共同决定。脑毛细血管表面积和脉络丛表面积分别为BBB 与BCSFB 内药物可通透表面积(surface area,S)。在PBPK 模型中,药物在屏障上的被动扩散速率可以用膜的渗透性-表面积(permeability-surface area product,PS)来描述。
在上述2 种屏障中还存在多种摄取或外排转运体,它们分布在屏障的两侧(见图1)。主动转运过程根据药物运输方向可以分为摄取转运和外排转运。帮助药物或内源性物质进入大脑的转运体称为摄取转运体,而将化合物移出大脑的转运体为外排转运体。人BBB 上活跃的转运蛋白包括有机阴离子转运多肽1A2(organic anion transporting polypeptide 1A2,OATP1A2)、有机阴离子转运蛋白3(organic anion transporters 3,OAT3)、单羧基反式转运蛋白1(monocarboxylate transporter 1,MCT1)、P-糖蛋白(P-glycoprotein,P-gp)、乳腺癌耐药蛋白(breast cancer resistance proteins,BCRPs)和多药耐药相关蛋白(multidrug resistance associated proteins,MRPs)。BBB 上大多数外排转运体位于血液侧,而BCSFB 上BCRP 和P-gp 位于CSF 侧,MRP 位于血液侧。
1.3 药物与蛋白质的结合
在脑组织中,药物与脑内蛋白质的结合可分为特异性结合和非特异性结合。特异性结合指药物与靶点的结合,这是发挥药效的基础。靶点可以是受体、酶、转运蛋白或离子通道。药物靶点根据其存在的空间位置可分为细胞外靶点或细胞内靶点,细胞内靶点可位于细胞质或细胞核内。在PBPK 模型中,药物与靶点的结合速率用kon表示,药物-靶点复合物的解离速率用koff表示,药物与受体的亲和力用Kd表示。药物与脑组织的非特异性结合包括药物与血浆蛋白及ECF 或CSF 中的蛋白质的结合。通常只有游离药物才能透过血管壁或生物膜。药物在脑内的非特异性结合会减少游离药物的扩散[10],从而影响药物在脑内进一步分布。在脑血管中,药物可能与血浆蛋白结合以便于运输,或结合后再解离从而发挥“药库”的作用。通常酸性药物与白蛋白结合,而碱性药物与α1-糖蛋白结合。此外,尽管ECF 与CSF 中蛋白质含量较低,这2 个体系中的药物-蛋白结合有时仍会被纳入考虑。药物与血浆或ECF、CSF 中的蛋白质结合后,结合型与游离型药物浓度处于动态平衡,游离型药物浓度与总药物浓度之间的比值被定义为药物的游离分数(unbound fraction,fu)。
2 脑组织生理药代动力学模型研究现状
脑组织PBPK 模型的建立基于脑组织的生理结构。脑组织的主要组成部分包括脑毛细血管、构成脑实质的各类细胞和ECF 以及CSF;此外还有将这些结构与循环系统隔离开的屏障——BBB 和BCSFB。然而,模型结构的确定并非直接将上述成分作为房室纳入,而是根据研究目的选择适用的最简单模型,因为越复杂的结构参数化越困难。本节综述了近年来文献报道过的脑组织PBPK 模型。PBPK 模型是建立在机体的生理、生化、解剖和药物热力学性质基础上的一种整体模型,它将解剖学上存在差异的器官或结构看作一个房室,房室间借助于血液循环或组织液流动来连接。脑组织PBPK模型中每个房室的建立依赖于两类参数:系统相关参数和药物相关参数。系统相关参数主要包括解剖学参数和生化参数,如组织液流速、组织大小、转运体或代谢酶活性等;药物相关参数包括药物热力学性质和药物与机体相互作用,如脂溶性、电离性、膜通透性、药物游离分数等。有文献报道了人、大鼠、小鼠、猴子和狗的CNS 生理参数[11],为脑组织PBPK 模型的建立提供了便利。
在全身PBPK 模型中,脑组织被视为一整个房室,通过血液循环与系统药代动力学(pharmacokinetics,PK)连接起来[12-14]。这类模型可以用于药物物料平衡的研究;也可以和药物毒性数据结合来探究药物的不良反应。全身PBPK 模型可以预测脑组织中整体药物浓度,但药物在脑组织中具体分布情况无法得知。此外,全身PBPK 模型的建立与验证需要来自多个组织中的药物浓度信息,然而实际研究中可能只关心脑组织中药物浓度,因此缺少其他组织的药物浓度数据,此时全身PBPK模型的应用存在限制。
因此,为了进一步探究药物在脑组织中的分布情况,结合微透析技术建立脑组织PBPK 模型的方法应运而生。使用脑内微透析技术来测定脑ECF 中游离药物浓度,可以将全身PBPK 模型中的脑室根据细胞内和细胞外空间进一步区分为单独的房室。微透析技术的一个显著优点是可以频繁地收集透析液样品进行PK 特征分析,因为ECF 中的液体实际上没有减少[15]。Tunblad 等[16]应用脑内微透析技术探究了建模时可使用的数据对模型结果的影响(见图2)。他们建立的模型包括动脉室、静脉室、外周室和脑细胞外液室。系统PK 参数通过动脉血中总浓度获得,而药物与蛋白结合率可以通过静脉血和ECF 中游离药物浓度计算得到。图3 给出了以吗啡为模型药物,应用这一模型对脑透析液、静脉透析液和动脉血中总药物浓度的观测数据、群体预测和个体预测。
Westerhout 等[17]2012 年报道的研究包含4 个CSF 房室的大鼠脑组织PBPK 模型,目的是探究不同的CSF 采样点是否在大鼠体内产生类似的PK 特征,从而推断人CSF 药物浓度是否可以代替ECF的药物浓度。模型结构见图4,在他们的模型中包含中央室、外周室、脑实质室和4 个不同的CSF 室。药物从脑ECF 转运至CSF 并依次经过LV、第3 和第4 脑室(third and fourth ventricle,TFV)、CM 和SAS。然而,由于缺少人脑内的药物浓度数据,他们对人脑内药物浓度的预测还有待临床数据验证。Yamamoto 等[18]改进了这一模型,并使用临床数据和文献中的数据[19]进行验证(见图5[20])。之后,他们又加入了亚细胞室、脑微血管室,并将BBB 和BCSFB 处的被动扩散分解为跨细胞和细胞旁2 个途径[21]。这一系列研究的目的是建立对不同药物都具有适用性的综合PBPK 模型,无需体内数据,根据生理和病理条件下系统特异性和药物特异性参数即可预测CNS 中多个区域的药物浓度-时间分布。
目前已报道的大部分脑组织PBPK 模型都是根据脑血管、CSF、ECF、细胞实质等脑组织成分区分脑室的,这些模型可以探究药物在BBB 上的转运情况但不能反映药物在依据结构划分的各个脑区(皮质、海马等)的分布情况。Zakaria 等[22]在2018 年提出了根据生理学结构区分脑室的大鼠CNS模型。这一模型内嵌于一个全身PBPK 模型中,并且包含5 个房室(见图6):脑血管、CSF、海马区(hippocampus,HP)、前额叶皮质区(frontal cortex,FC)和脑组织剩余部分(rest of brain,ROB),模型结构使用文献报道过的苯妥英[23]和卡马西平[24]在不同脑区中的数据进行验证(见图7)。这一模型也被用于预测了人脑内不同区域的药物PK特征。尽管缺乏临床数据验证,这种根据脑区划分方式的方法仍然是一个很有意义的尝试,因为实验表明许多靶向CNS 的药物在脑内的分布存在区域特异性[23,25],而这与脑组织不同结构的解剖学差异和功能有关。

图2 基于微透析数据建立的脑组织生理药代动力学模型[16]Figure 2 Physiologically-based pharmacokinetic model of the brain tissue based on microdialysis data[16]
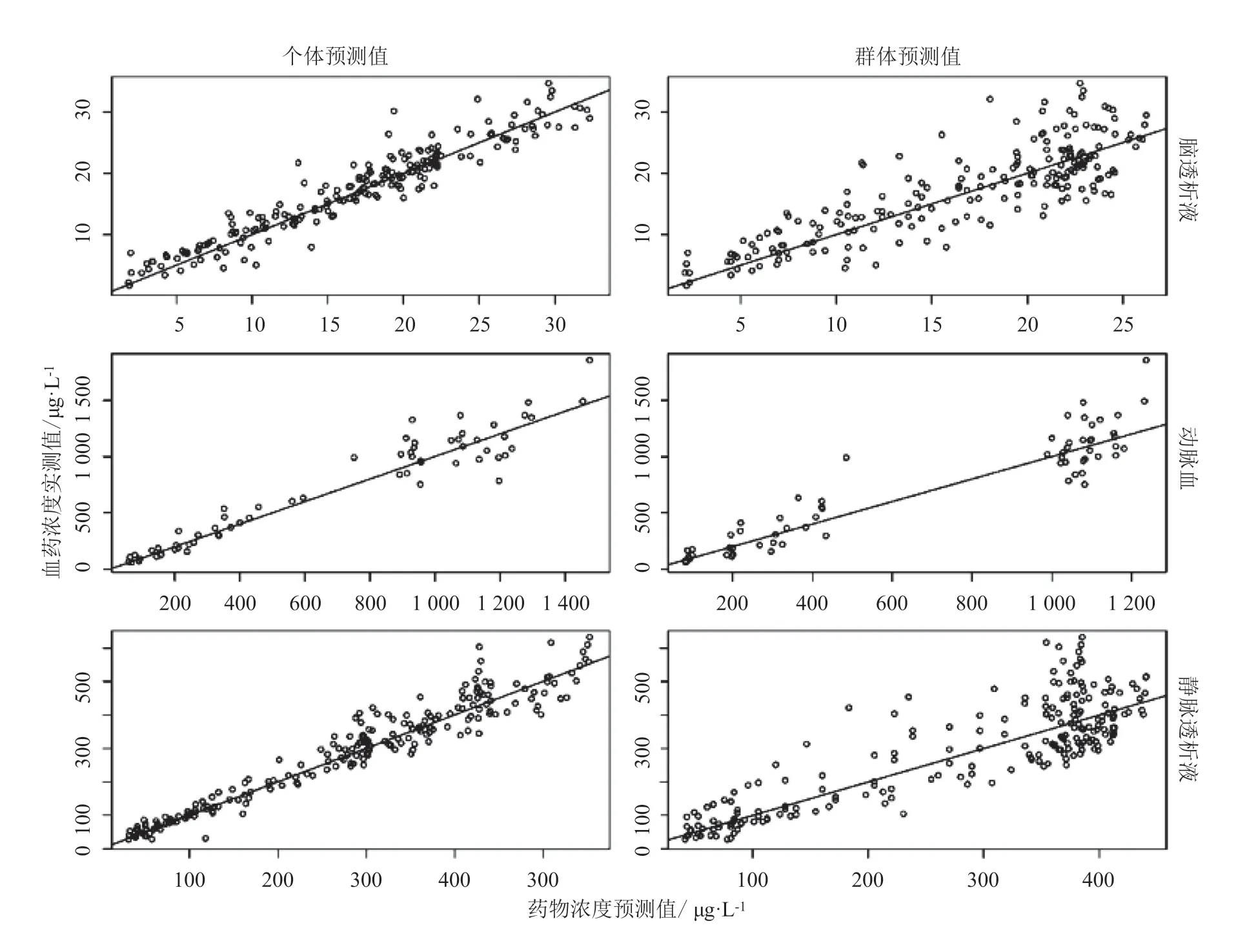
图3 脑透析液、静脉透析液和动脉血中吗啡浓度的实测值-群体预测值和实测值-个体预测值散点图[16]Figure 3 Scatter plots of the observed data,population predictions and individual predictions of morphine concentrations in cerebral dialysate,venous dialysate,and arterial blood [16]
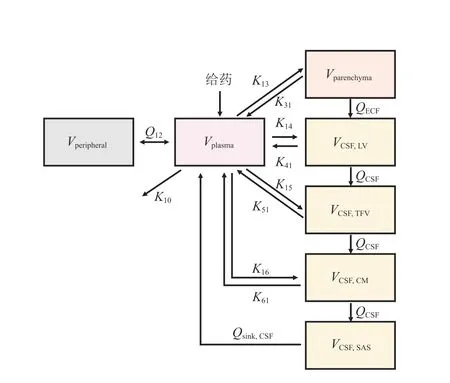
图4 包含4 个脑脊液房室的脑组织生理药代动力学模型结构示意图[17]Figure 4 Physiologically-based pharmacokinetic model of the brain tissue containing four cerebrospinal fluid chambers [17]
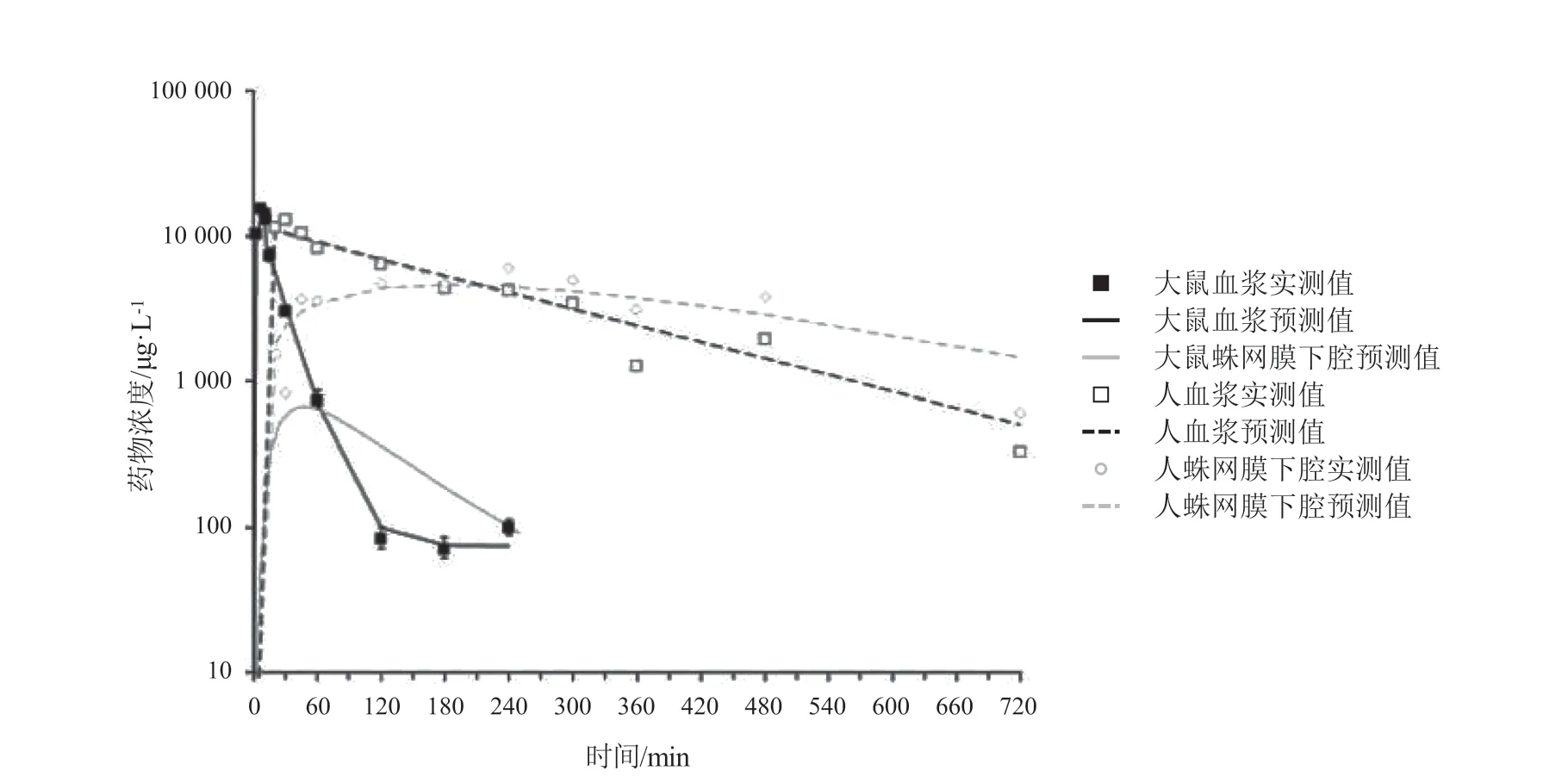
图5 大鼠和人静脉注射15 mg·kg-1 对乙酰氨基酚后血浆和蛛网膜下腔中药物浓度预测值与实测值[17,20]Figure 5 Observed data and predictions of drug concentrations in plasma and subarachnoid space in rat and human after i.v.infusion of 15 mg·kg-1 acetaminophen [17,20]
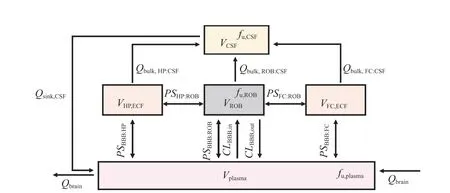
图6 根据生理学结构区分脑室的大鼠脑组织生理药代动力学模型[22]Figure 6 A region-specific rat brain physiologically-based pharmacokinetic model established according to the physiological structure of the brain tissue [22]
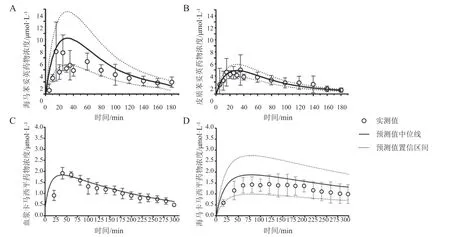
图7 大鼠给予苯妥英或卡马西平后脑区或血浆中药物浓度预测值与实测值[22]Figure 7 Observed data and predictions of drug concentrations in rat plasma and brain regional compartments after phenytoin or carbamazepine administration [22]
总的来说,本节所述的PBPK 模型为分析研发中药物在BBB 和BCSFB 上的转运提供了良好的基础,同时也为预测人体内未结合的ECF 药物浓度提供了合理的生理学方法。然而,这一类模型在进行种属间转化时仍然存在限制。尽管固定了系统相关参数,模型中的药物相关参数仍然是通过数据拟合得到的。尤其是描述药物跨BBB 或BCSFB 转运过程的参数往往代表着几种动力学过程的净效应,将模型中系统相关参数替换为人的生理值的种属间转化方法需假设不同物种间跨BBB 和BCSFB 转运具有类似的机制,而这显然是不成立的。
3 脑组织生理药代动力学模型的应用
3.1 基于机制的生理药代动力学/药效学模型
脑组织PBPK 模型不仅可以用于估计BBB 和BCSFB 的流入和流出参数来解释药物的脑内PK 特征,还能为脑ECF浓度与药效学(pharmacodynamics,PD)数据建立联系。Hammarlund-Udenaes 及其同事基于大鼠脑内微透析数据和阿片类药物的镇痛作用,报道了一系列基于机制的PBPK/PD 模型[26-30]。他们的研究目的之一是定量解释吗啡和羟考酮的PK/PD关系[30]。这2 种镇痛药在体外实验中对阿片受体的亲和力有显著差异,但在大鼠血浆内药物浓度相近时可以产生等效的镇痛效应。他们认为这可能与2种药物的BBB 转运差异有关。基于机制的PBPK/PD 模型模拟出的药物浓度结果支持这一假设,BBB对羟考酮的摄取速率高于外排速率,而吗啡的结果相反,这使得羟考酮的脑内ECF 游离药物浓度比吗啡高6 倍[29-30]。这一结果提示,在研究靶点位于CNS 的药物时,应考察药物的脑内游离药物浓度与药物效应的PK/PD 关系,而不是血浆PK/PD 关系。
此外,脑组织的PK 还可以和受体占有率数据(receptor occupancy,RO)结合,建立PBPK/RO 模型。Wong 等[31]开发了2 种常用的抗抑郁药氯氮平和利培酮的大鼠和人的全身PBPK/RO 模型,充分预测了人体内原药和代谢物的血浆药物浓度(见图8)及其对人脑内多巴胺D2受体的占有率水平(见图9)。PBPK/RO 模型的应用可以从D2受体扩展到CNS 的其他受体,从而评价药物的受体选择性。所有已上市的抗精神病药物对CNS 中的非D2受体都有明显的亲和力[32],而它们与5-羟色胺(5-HT)受体或乙酰胆碱受体的相互作用是引起CNS 不良反应的重要原因。例如,氯氮平引起的代谢紊乱(如体质量增加或糖尿病)可能与其对组胺H1受体和5-HT2C受体的过度亲和力有关[33];而氯氮平对5-HT2A受体和毒蕈碱M1的高亲和力可能诱发运动障碍[34]。因此,通过PBPK/RO 模型评价药物的受体选择性具有重要的临床意义。
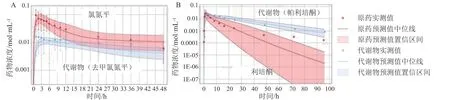
图8 人口服氯氮平和利培酮给药后血浆中原药和代谢物浓度预测值与实测值[31,35-36]Figure 8 Observed data and predictions of parent drug concentrations and metabolite concentrations in plasma after oral administration of clozapine and risperidone in human [31,35-36]
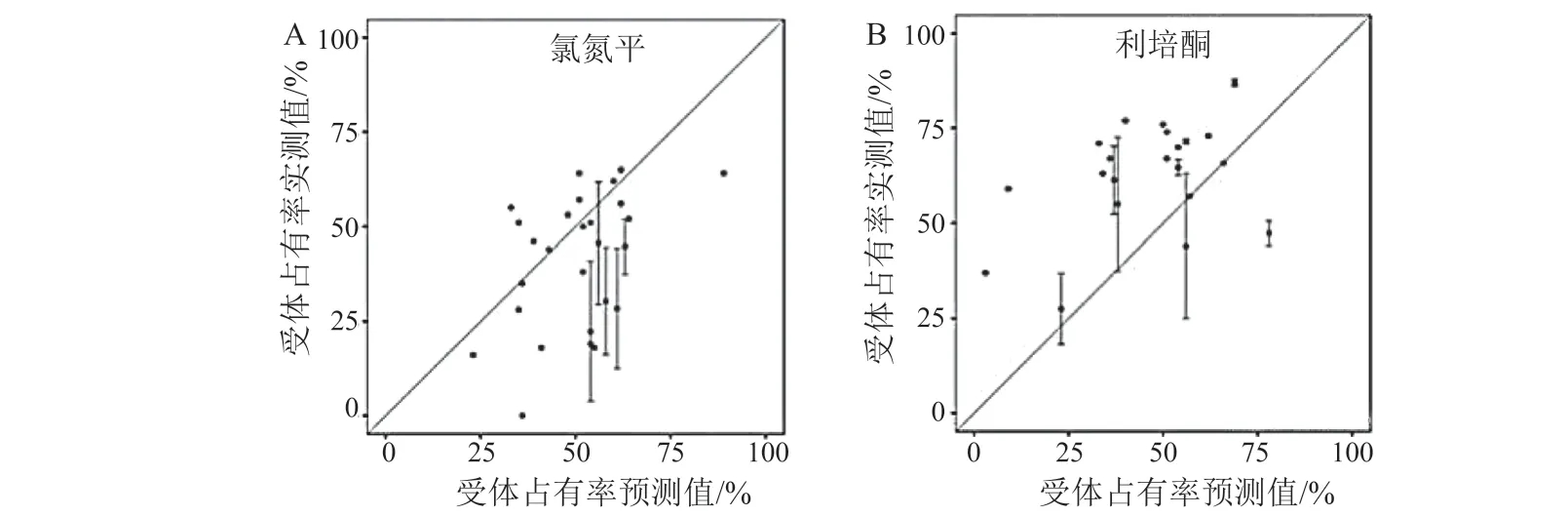
图9 人口服氯氮平和利培酮后D2 受体占有率的预测值与实测值[31]Figure 9 Observed data and predictions of D2 receptor occupancy after oral administration of clozapine and risperidone in human[31]
3.2 体外-体内外推/生理药代动力学模型
PBPK 模型中的系统相关参数具有实际的生理意义,部分参数可在机体内直接测得,从而为这一类模型的种属间转化提供了良好的基础。除了直接将动物模型中的系统相关参数替换为人的生理值外,近年来越来越多的研究者在估计PBPK 模型的参数时采取了基于体外-体内外推(in vitro-in vivoextrapolation,IVIVE)的策略。这种“自下而上(bottom-up)”的方法是将体外实验测得的转运体或代谢酶的动力学参数,通过比放因子(scaling factor,SF)转化为PBPK 模型中的参数。SF 表示药物在体外实验及体内环境中测得的动力学过程之间的活性差异[37]。此外,还要考虑体内与体外实验体系中转运体的蛋白表达水平差异。
Ball 等[38]在2012 年提出的模型中,将脑组织分为脑血管和脑实质2 个房室,并内嵌于大鼠全身PBPK 模型中。他们通过体外实验测定了BBB 对吗啡和羟考酮的被动转运或主动转运通透性,之后将模型中的BBB 通透性(P)固定为测量值,应用PBPK 模型模拟了大鼠脑内的游离药物浓度(见图10)。他们的模型中用相对活性因子(relative activity factor,RAF)表示转运蛋白在体外-体内活性的差异,这一参数值可以由模型与大鼠观测值拟合得到。在大鼠到人的转化时,他们将模型中大鼠的生理参数替换为人体的生理值,并使用了与大鼠模型中相同的RAF。他们假设在大鼠和人之间,每单位膜表面积或每单位微血管蛋白量的相关转运体的表达水平和活性相同。2014 年,Ball 等[39]提出了包含脑血管、CSF、ECF 和细胞实质4 个房室的脑组织PBPK 模型,并详细讨论了在临床前研究阶段建立脑组织IVIVE-PBPK 模型的策略以及参数化方法。

图10 大鼠静脉推注吗啡和羟考酮后脑内游离药物浓度的实测值与预测值[38]Figure 10 Observed data and predictions of unbound brain concentrations after i.v.infusion of morphine and oxycodone [38]
Lu 等[40]在2016 年提出了用于描述人脑内药物处置的4Brain 模型。该模型在一项0 期临床试验中被用来预测一个研发中的药物AZD1775 穿透BBB的能力[41]。作者通过文献中报道的对乙酰氨基酚和苯妥英的临床数据验证了4Brain 模型。这2 个药物分别代表不同的脑内转运机制,对乙酰氨基酚进入脑组织主要依靠被动转运[42],而苯妥英作为P-gp的底物,主要通过转运体介导的主动转运进入脑组织中(见图11)[43]。在4Brain 模型中,基于IVIVE策略由体外实验得到的药物相关参数包括:1)BBB与BCSFB 的PS;2)BBB 与BCSFB 上转运体活性(CLin/CLout);3) 脑内药物的代谢清除率(CLmet)。其他系统相关参数由文献中报道过的生理值得到。然而,这一模型没有纳入ECF,因而不能预测作用于膜结合受体的药物的受体结合动力学以及药物的效应[18,40]。4Brain 模型用于预测对乙酰氨基酚给药后,虚拟人群血浆和CSF 中药物浓度(见图12)。
4 结语
脑组织PBPK 模型的建立基于脑组织的生理、生化、解剖和药物热力学性质。相较于房室PK 模型,脑组织PBPK 模型为种属间转化提供了更好的基础。在新药研发中,脑组织PBPK 模型可以用于预测药物在脑内的PK 行为、探究药物的PK/PD 关系,或基于IVIVE 策略进行种属间转化。随着对脑组织解剖学研究的不断深入以及微透析技术的发展,模型结构和参数越来越接近真实的生理情况,这有助于提高模型的预测准确性。总之,基于脑组织PBPK 模型预测药物在人脑内的转运和分布情况可以成为新药研发中的重要策略,这使得这一研究领域具有良好的应用前景。
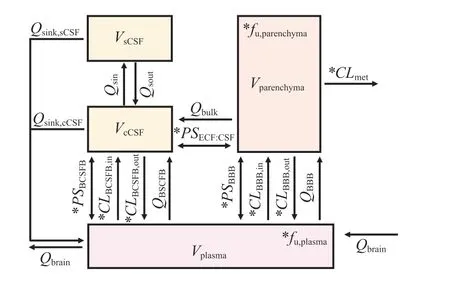
图11 基于体外-体内外推方法建立的4Brain 模型结构[40]Figure 11 Model structure of the 4Brain model using the in vitro-in vivo extrapolation approach [40]
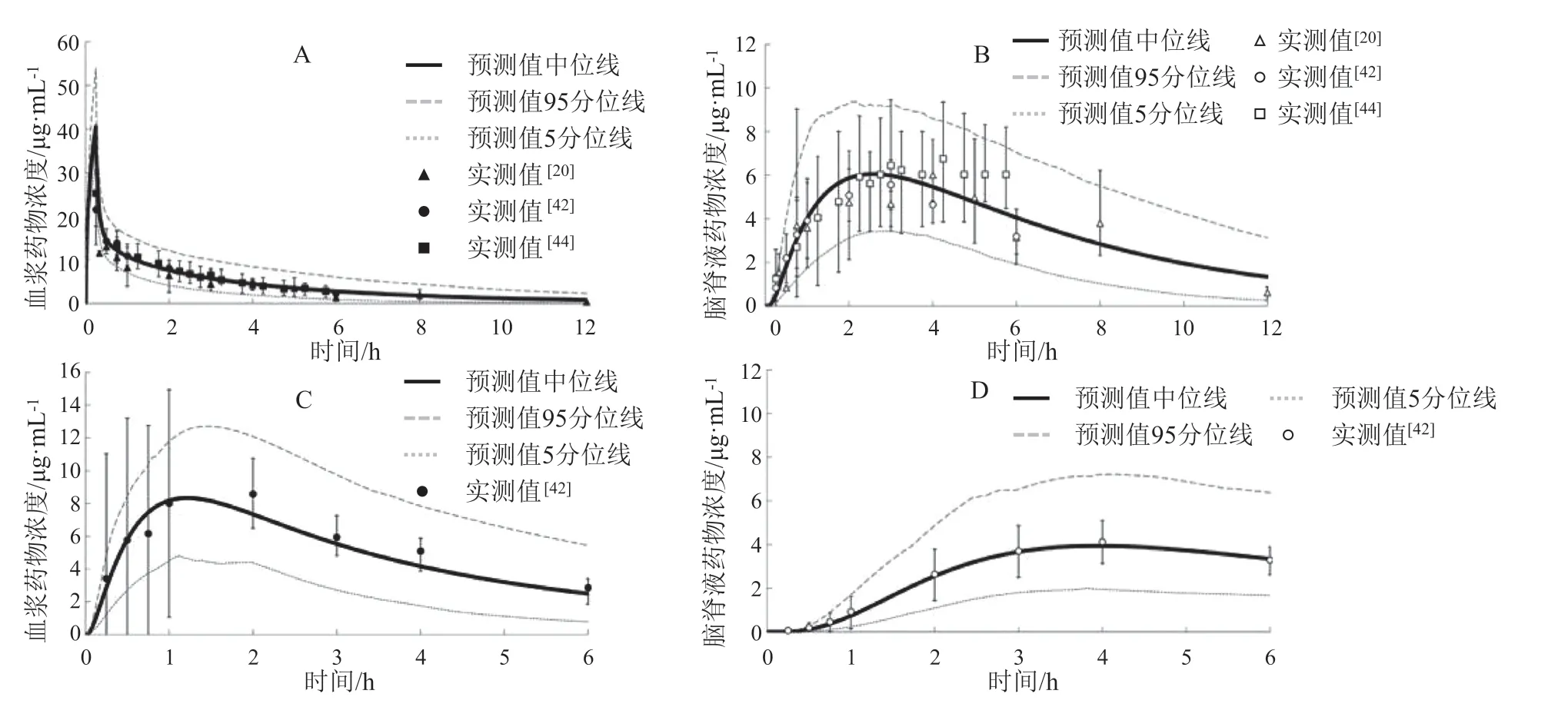
图12 对乙酰氨基酚给药1 000 mg 后药物浓度预测值与实测值[20,40,42,44]Figure 12 Observed data and predictions of drug concentrations after administration of 1 000 mg paracetamol [20,40,42,44]
猜你喜欢
江苏安全生产(2022年8期)2022-11-01
小资CHIC!ELEGANCE(2021年36期)2021-10-15
中国药剂学杂志(网络版)(2021年1期)2021-02-24
艺术评鉴(2020年5期)2020-04-30
音乐研究(2019年5期)2019-11-22
湖南饲料(2019年4期)2019-10-17
中国生物医学工程学报(2019年5期)2019-07-16
中国人兽共患病学报(2018年10期)2018-12-08
人大建设(2018年10期)2018-12-07
中国当代医药(2018年21期)2018-11-10
