
多小穗种质10-A穗部性状的QTL定位与分析
2020-02-26 01:04张志鹏赵晓芳易晓余赵梦梦沈婧玥蒲至恩陈国跃
西南农业学报 2020年10期
张志鹏,赵晓芳,易晓余,丁 丽 ,赵梦梦,沈婧玥,蒲至恩,陈国跃,李 伟*
(1. 四川农业大学小麦研究所,四川 成都 611130;2. 四川农业大学农学院,四川 成都 611130)
【研究意义】普通小麦(TriticumaestivumL., 2n = 42, AABBDD)由A、B、D 3个基因组组成,基因组庞大(约14 500 Mbp),是世界上重要的经济和粮食作物之一[1-3]。中国是小麦生产和消费大国,据统计,目前我国小麦的年消费总量在1.07~1.09×108t,其产量对中国的粮食安全有着重要影响[4-6]。单位面积穗数、穗粒数和千粒重是决定小麦单产高低的关键因素。因此,小麦穗部性状的表现对小麦产量的最终形成具有重要影响。【前人研究进展】近年来,研究人员对小麦的穗部性状进行多方面研究。Li等[7]利用重组自交系群体(RIL)对农艺性状进行QTL定位,可解释4.42 %~70.25 %的表型变异;张坤普等[8]利用双单倍体群体(DH)进行穗部性状的QTL定位,表型变异的贡献率为1.48 %~15.63 %;Cui等[9]利用重组自交系(RIL)检测到控制穗部性状的QTL,可解释表型变异率为4.94 %~10.97 %。因此,通过对小麦穗部性状进行基因定位,对提高相关性状的育种选择效率,促进优异种质在小麦育种中的应用具有重要意义。DArT(Diversity Arrays Technology)即多样性微阵列技术,是一种以限制性酶切为基础、依赖于芯片杂交来区分基因组中座位多态性差异的新型分子标记技术[10-11]。DArT 标记已用于构建大麦、硬粒小麦等作物的遗传图谱[12-13]。在普通小麦中,Huynh 等[14]利用SSR和DArT 标记相结合的方法构建了小麦DH 群体遗传图谱, 并对该群体进行了小麦籽粒果聚糖含量QTL分析;Griffiths 等[15]利用DArT 标记和SSR标记构建了4个双单倍体(doubled haploid, DH)群体的遗传连锁图谱。曹东等[16]利用DArTseq标记在F2群体进行株高、穗长、穗粒数和千粒重等农艺性状的QTL定位,可解释的表型变异率为1.4 %~29.8 %。小麦种质10-A是四川农业大小麦研究所创制的含有黑麦血缘的小穗数超过26个的不分枝多小穗数品系[17],染色体定位表明其单株籽粒产量受5A、7A、1B、2B、6B、2D 和7D等染色体上的基因所控制[18]。以10-A与遗传差异较大的材料配制的组合分析表明,小穗数、抽穗期、穗粒数和粒重等性状在不同的基因效应上存在着较大的差异,加性、显性和上位性各分量对性状在世代间的遗传差异均具有不可忽视的重要贡献[19]。对10-A在不同播期下的农艺性状评价表明,播期对多小穗品系10-A的多小穗数影响不明显[20]。10-A的多小穗数目的增加与植株发育阶段有关,增长单棱期和二棱期可显著增加小穗数的数目[21]。利用10-A杂种对小穗的幼穗发育分析表明,其后代多小穗的产生是继承了10-A较高的分化速率,而与分化期并没有显著关系[22]。【本研究切入点】本研究利用多小穗小麦种质10-A与BE89杂交获得的含有186个单株的F2群体及其衍生的 F2∶3家系群体,采用DArT标记构建了小麦分子遗传图谱,并利用4个环境条件下的表型数据对穗部性状进行QTL定位。【拟解决的关键问题】为育种中合理利用10-A等材料提供参考依据,以及为小麦穗部性状的精细定位、基因克隆和分子标记辅助选择育种提供理论基础。
1 材料与方法
1.1 试验材料
多小穗种质10-A具有多花多粒、大穗、小穗数多、千粒重低等特点[18-19];小麦品系BE89为 Batavia/Erine杂交组合选育的寡小穗数稳定品系,具有芒长、穗短、小穗数少等特点。2015年4月利用10-A(母本)与BE89(父本)进行杂交获得种子,2015年10月下旬播种F1代,成熟后混合收获种子,2016年10月播种上一年所收获种子,形成由186株组成的F2群体,成熟时按单株脱粒装袋,每单株收获种子等分成3份,2017年10月下旬种植F2收获的单株种子,形成株系,组成F2∶3群体。所有供试材料均由四川农业大学小麦研究所提供。
1.2 试验方法
1.2.1 田间试验和性状调查 田间试验在四川农业大学温江、崇州、雅安教学基地开展。2016年10月下旬将10-A/BE89 F2群体与亲本种植于四川农业大学温江(E4)环境;2017年10月下旬将10-A/BE89 F2∶3家系和亲本分别种植于温江(E1)、崇州(E2)和雅安(E3),每个系种植2行。田间播种均采用单粒点播,行长2 m,株距0.1 m,行距0.3 m;田间管理参照大田管理方式,生长期间未发生严重病虫害和倒伏现象。
灌浆后期田间调查穗长(Spike Length,SL)、芒长(Awn Length,AL)、总小穗数(Total Spikelet per Spike,TSS),成熟后收获室内脱粒调查穗粒数(Kernel Number per Spike,KNS)和千粒重(Thousand-Kernel Weight,TKW)。其中,F2群体调查单株,F2∶3群体每个株系中间随机调查3株,取平均值。性状调查标准参照李立会和李秀全所著[23]《小麦种质资源描述规范和数据标准》。
1.2.2 DArT标记检测 在F2群体幼苗四叶期时,取10-A/BE89 F2群体单株叶片 2 g 左右,置于液氮中备用。采取CTAB法[24]提取小麦的基因组 DNA,并用1 %的琼脂糖电泳检测DNA的质量,用核酸测定仪(NanoDrop 2000/2000c)测定DNA的浓度与纯度。
高通量 DArT 标记检测由澳大利亚 Diversity Arrays Technology Pty Ltd分析后提供,产品为 Wheat GBS1.0 芯片,分析检测方法参照 Wenzl等[13]的报道。
1.2.3 表型数据分析 利用 Microsoft Excel 2010 软件对表型数据进行整理;利用 IBM SPSS Statistics 20.0对群体的性状表型值进行描述性统计分析;利用SAS 9.1.3软件计算最佳线性无偏估计值(BLUP)。
1.2.4 连锁图谱的构建和QTL分析 筛选双亲之间的差异标记,再将其按照孟德尔分离理论比率(1∶3)进行卡方检验,除去缺失率 > 5 % 和偏分离的标记,最后所得标记用于遗传连锁图谱的构建;利用JoinMap 4.0进行连锁分析,构建遗传连锁图谱。
使用IciMapping4.0进行QTL定位,同时计算各QTL的加性效应和贡献率。作图方法采用完备区间作图法(ICIM),PIN 设置为1,步长为1 cM,设定模拟运算(permutation)1000 次为标准,确定最终的LOD 阈值,并以此确定QTL在染色体上的位置及数目。
2 结果与分析
2.1 群体的性状表现
对亲本10-A和BE89,以及双亲组配的F2和F2∶3群体在4个环境下的穗长、芒长、总小穗数、穗粒数和千粒重5个穗部性状进行了方差分析(表1)。结果表明,10-A在4个环境中均能保持总小穗数超过28个的多小穗特性,在穗长、穗粒数、总小穗数、芒长和千粒重数值上与父本BE89差异极显著。穗部性状的均值在不同环境中表现出差异。穗长(SL)平均数在E1中最高为11.08 cm,在E2中最低为10.18 cm;芒长(AL)平均值在E3中最高为3.52 cm,在E4中最低为3.20 cm;总小穗数(TSS)平均数在E1、E3中最高为22.1个,在E2中最低为20.2个;穗粒数(KNS)平均数在E1中最高为35.6粒,在E2中最低为23.8粒;千粒重(TKW)均值在E1中最高为41.82 g,在E2中最低为27.77 g。群体中所有性状的平均变异系数为27.88 %。其中,芒长在4个环境下的平均变异系数最大,为44.51 %;总小穗数的平均变异系数最小,为13.04 %。穗长在E1中平均变异系数最高为25.7 %,最小在E2为12.5 %;芒长在E3中平均变异系数最高为45.7 %,最小是E1为42.5 %;总小穗数在E4中平均变异系数最高为14.9 %,最小在E3为11.6 %;穗粒数在E4中平均变异系数最高为44.3 %,最小在E1为32.1 %;千粒重在E2中平均变异系数最高为35.0 %,最小在E1为20.0 %。穗长、芒长、总小穗数、穗粒数和千粒重的遗传力均较高,分别为0.95、0.86、0.90、0.94和0.96。利用最佳线性无偏预测值(BLUP)做各性状的正态性检验,5个穗部性状在4个环境下均表现连续变异,基本呈正态分布,表现为数量性状遗传。性状均表现出不同程度双向超亲分离现象,表明控制相关性状的优势等位基因随机分布于双亲的染色体上[25]。
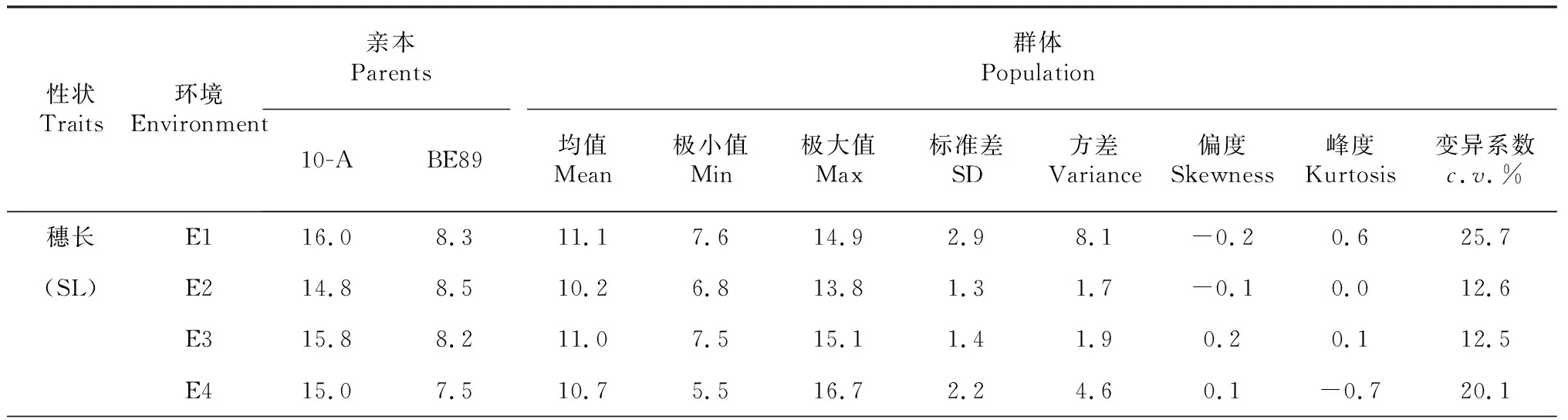
表1 群体及其亲本的性状表现
2.2 分子遗传图谱构建
双亲之间共筛选到4058个差异DArT标记,经卡方检验除去缺失率>5 % 和偏分离的标记后,最后共有 968 个DArT标记可用于遗传连锁图谱的构建。所有标记成功连锁到小麦21条染色体上,构建的遗传图谱的总长度为1878.408 cM(表2),每条染色体的平均长度为89.448 cM,标记间平均遗传距离1.94 cM。其中1B染色体上的标记数目最多,为197个,遗传图谱上染色体也最长(224.337 cM),标记间的平均遗传距离为1.138 cM;染色体标记数目其次的是2B染色体,有136个;标记数量最少是1D、7A和7B 3条染色体,均只有8个标记,其中7B染色体标记长度最短(10.624 cM),平均遗传距离为1.33 cM。染色体上标记间的平均遗传距离最短是4A染色体,有75个标记,平均遗传距离为0.63 cM;平均遗传距离最长是7A染色体,为5.48 cM。

续表1 Continued table 1

表2 基于F2群体构建的DArTs标记的遗传连锁图谱
2.3 QTL定位分析
采用复合区间作图法,5个穗部性状共定位到15个贡献率大于10 %的 QTL 位点。在 F2群体中检测到7个QTL位点,在F2∶3群体中检测到8个QTL位点,这些QTL位点分布在1A、1B、2A、2D、3B、5D和6B等 7条染色体上(表3和图1),单个QTL位点可以解释表型变异为7.64 %~21.80 %。其中,有6个QTL位点集中分布在2A染色体上,而3B和6B染色体上各检测到1个QTL位点。
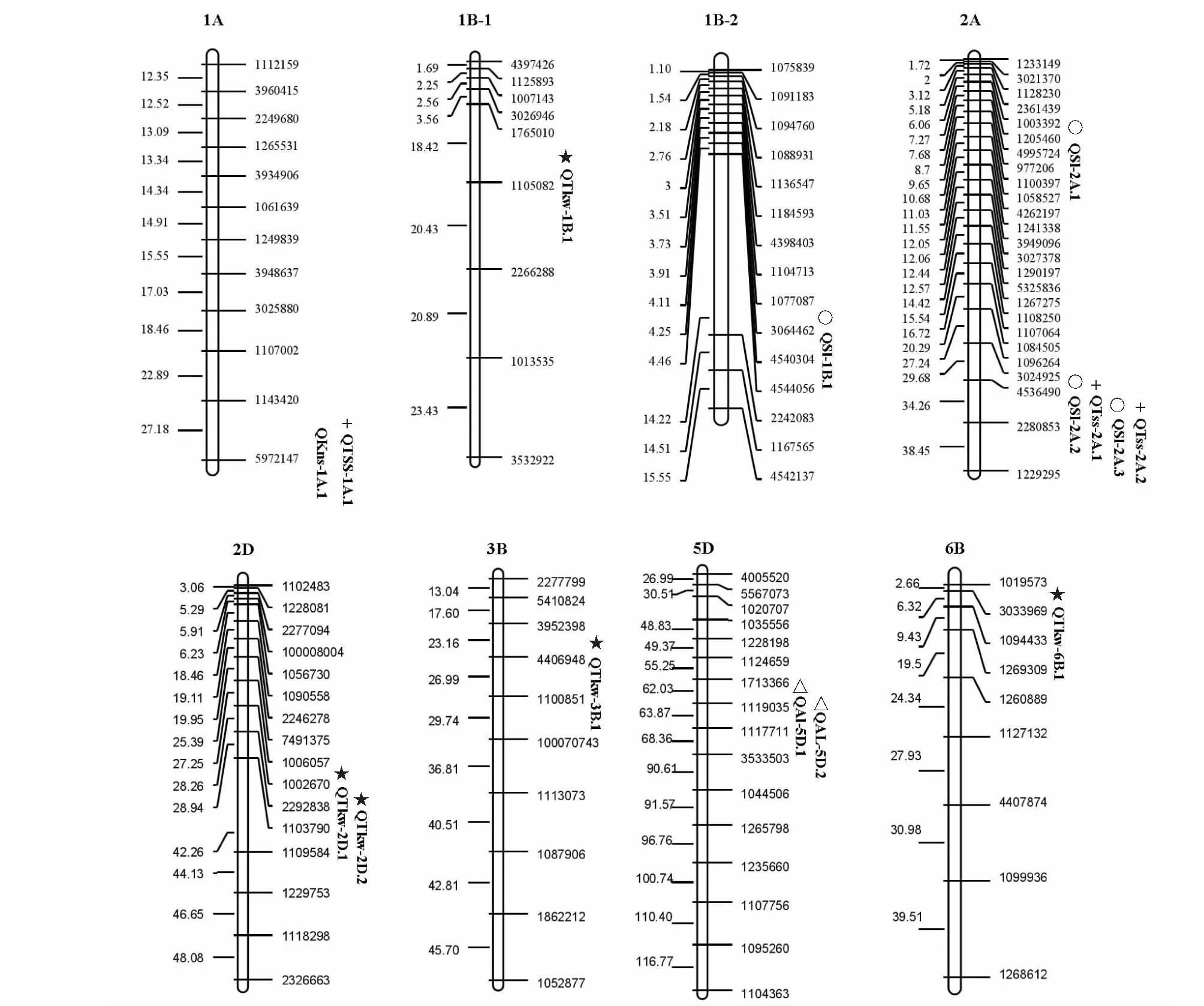
不同形状代表不同的性状的QTL;○代表穗长;△代表芒长;+代表总小穗数;☆代表穗粒数;★代表千粒重Different shape represent different QTLs for different traits;○stands for spike length;△stands for awn length;+stands for total spikelet per spike;☆stands for kernel number per spike;★stands for 1 thousand-kernel weight

表3 穗部性状QTL定位结果
2.3.1 穗长的QTL 共检测到4个控制穗长的QTL位点,定位在1B和2A染色体上,其中F2群体中检测到的QSl-2A.1、QSl-2A.2和QSl-2A.3,可以解释的表型变异率分别为12.17 %、21.80 %和20.24 %;QSl-1B.1在F2∶3群体中检测到,可以解释的表型变异率10.31 %。QSl-2A.1位点的增效基因来自于母本10-A,而QSl-1B.1、QSl-2A.2和QSl-2A.3位点的增效基因来自于BE89。
2.3.2 芒长的QTL 2个与芒长相关的QTL定位在5D染色体上,其中QAl-5D.1位点在 F2群体和F2∶3群体中均检测到,对表型变异的贡献率分别为7.64 %和13.81 %。所检测到的QTL的加性效应均为正值,增效基因均来自母本10-A,对芒长表现出抑制作用。
2.3.3 总小穗数的QTL 4个与总小穗数相关的QTL被定位在1A和2A染色体上,单个QTL可解释11.58 %~15.37 %的表型变异。其中,QTss-1A.1位点在F2∶3群体检测到,解释13.54 %的表型变异;QTss-2A.1和QTss-2A.1位点只在F2群体中被检测到,分别解释11.58 %与15.37 %的表型变异率;所有QTL位点的加性效应均为正值,其增效效应均来自母本10-A,表明10-A能促进后代的小穗数提高。
2.3.4 穗粒数的QTL 1个与穗粒数相关的QTLQKns-1A.1被定位在1A染色体上,可解释11.18 %的表型变异,在F2∶3群体中检测到,且增效基因来源于母本10-A,与10-A穗粒数多的特性相一致。
2.3.5 千粒重QTL 5个控制千粒重的QTL被定位在1B、2D、3B、3D和6B染色体上,单个QTL可以解释10.91 %~19.09 %的表型变异,QTkw-1B.1解释的表型变异率最高为19.10 %。其中,4个QTL位点在F2∶3群体中检测到,1个QTL位点在F2群体中检测到。千粒重的所有QTL的增效基因来源于父本BE89。
3 讨 论
本研究利用多小穗10-A与BE89构建的F2和F2∶3群体,采用DArT标记,在4个环境中共定位到控制穗长、芒长、总小穗数、穗粒数和千粒重等穗部性状的15个QTL位点,分布在7条染色体上,单个QTL位点可解释表型变异为7.64 %~21.80 %。其中,控制芒长的QTL位点QAl-5D.1在F2和F2∶3群体中均能检测到,其他QTL位点均只能在单一群体环境中检测到在,表明这些性状很大程度上受到了基因型和环境的双重影响。而穗长、芒长、总小穗数、穗粒数和千粒重的遗传力均超过0.85,表明遗传因素是主要因素。在所检测到的QTL位点中,有2个QTL对表型的贡献率超过20 %,为重要主效QTL位点。本研究中检测到的控制穗粒数(QKns-1A.1)和千粒重(QTkw-6B.1)等位点与前人的定位结果相一致[26-36],而穗长、芒长、总小穗数和千粒重的部分定位结果尚未见报道,是本研究新发现的QTL。
郑有良等[26]对控制穗长的基因进行染色体定位发现,10-A的穗长受1B、2D、3D、5A、6D和7A等染色体上的基因所控制;Deng等[27]在2A、2D、3A、4B、4D、5D、6B 和 7B染色体上定位11个控制穗长的QTL位点。本研究中穗长QTL定位在1B和2A染色体上,与前人定位在同一染色体上。在2A染色体上检测到了3个控制穗长的QTL位点,表明该染色体对穗长发育调控具有重要作用,但3个QTL位点间的关系还有待进一步的研究。
芒长由4 个主要的抑制基因和1 个促芒基因以及部分微效基因共同决定[28]。这些基因不同的组合决定芒的不同表现,基因B1、B2和B3为芒抑制基因,其中B1作用最强,B3作用最弱;A基因促进芒的伸长;Hd基因是产生钩芒的基因,也是芒抑制基因[29]。其中,Hd基因被定位在4AS染色体[30],B1基因被定位在5AL染色体上[31],B2基因被定位在6AL染色体[32]。也有研究表明,在5A、1B、3B、4B、5B和6B染色体上存在促进芒发育的基因,在2A、6A和2B染色体上存在抑制芒发育的基因[32]。本研究在5D染色体定位到控制芒长的QTL位点QAl-5D.1在2个环境中均可检测到,且均表现出对芒长的抑制作用,这2个QTL位点前人都未曾报道过,可能是新的芒长抑制基因位点。
Kuuar等[33]、Cui等[9]、Wang等[34]和Millet等[35]分别在2B、3D、5D和2D染色体上定位到控制总小穗数的QTL。本研究所检测到的控制总小穗数的QTss-1A.1、QTss-2A.1和QTss-2A.2位点前人均没有报道,推测为新的QTL位点。QTss-1A.1位点在2个环境中均能检测到,表现稳定,值得重点关注。小麦1A和2A染色体是控制总小穗数的QTL的热点区域,且在1A和2A染色体上鉴定到了新QTL位点,接下来会重点关注1A和2A染色体的控制总小穗数的QTL。所有控制小穗数的QTL位点的增效均来自10-A,表明10-A 是适用于改良育成品种的总小穗数目进而提高产量的优异种质。
Blanco等[36]将与穗粒数相关的QTL位点定位到1AL、2AS、2BL、4AL、4BS 和5AL等6个染色体上。本研究只在1A染色体上检测到主效QKns-1A.1位点,与前人定位染色体相同。
郑有良等[18]的染色体定位表明,10-A的千粒重受1B、2B、2D、5A、6B和7D等染色体上的基因所控制。Zhang等[37]对前人有关千粒重的QTL研究进行了综述,几乎全部小麦21染色体上都检测到了控制小麦千粒重的QTL,本研究利用DArT标记也在1B、2D和6B上检测到控制千粒重的主效QTL位点,与前人的染色体定位结果一致。此外,本研究检测到3B和3D染色体上存在新的QTL位点。
本研究所检测到的穗部性状QTL位点对表型变异的贡献率相对较低,这与前人的一些研究结果[26-37]类似,可能是因为这些性状大都属于数量性状,由微效多基因控制,且受环境影响较大,这些QTL位点的位置和效应有待于进一步检验。
4 结 论
本研究通过对F2群体和F2∶3群体多环境下的穗部性状进行表型鉴定,结合DArT分子标记定位到了15个与穗部性状相关的QTL位点。与前人研究比较后发现,这些QTL大多为新的位点,表明多小穗种质10-A是可用于育种应用的优异种质。本研究对于10-A穗部性状的初步定位结果,为其分子设计育种应用,以及控制穗部性状的位点/基因的进一步研究提供了理论基础。
猜你喜欢
分子催化(2022年1期)2022-11-02
中国现代医生(2022年21期)2022-08-22
烟草科技(2021年6期)2021-06-24
天津医科大学学报(2021年1期)2021-01-26
医药前沿(2020年20期)2020-11-10
三农资讯半月报(2020年2期)2020-03-09
科学之谜(2019年3期)2019-03-28
生物学教学(2018年4期)2018-11-29
电脑知识与技术(2018年19期)2018-11-01
科学之谜(2018年8期)2018-09-29
