
紫草总色素β-环糊精包合物的定性鉴别及左旋紫草素含量测定*
2020-02-11 07:58李俊江刘向娥李季文张增志牛建雄
中国药业 2020年3期
李俊江,刘向娥,李季文,张增志,牛建雄
(1.甘肃省中医院科研制剂中心,甘肃 兰州 730050; 2.甘肃省中医院白银分院,甘肃 白银 730900;3.甘肃陇中药业有限公司,甘肃 白银 730900)
紫草为紫草科植物新疆紫草Arnebia euchroma(Royle)Johnst.或内蒙紫草Arnebia guttataBunge的干燥根[1],主要活性成分为多种羟基萘醌类化合物(紫草羟基萘醌总色素)[2-3],药用活性广泛,但普通口服制剂的稳定性较差。β-环糊精包结紫草总色素可避免紫草总色素及衍生物长时间在光、空气和高温环境下产生聚合作用而失去活性[4-6],扩大临床使用范围。本研究中对紫草总色素β-环糊精包合物进行了定性鉴别与含量测定,建立了薄层色谱(TLC)鉴别及高效液相色谱(HPLC)含量测定方法。现报道如下。
1 仪器与试药
1.1 仪器
PerkinElmer A-10型高效液相色谱仪(美国PerkinElmer公司),包括717自动进样器,二极管阵列检测器(DAD);HS6150型超声波清洗仪(天津恒奥科技发展有限公司);Mettler AE240型电子分析天平(德国Sartorius赛多利斯公司,万分之一);HXGZ-000800型电热恒温干燥箱(连云港医疗器械设备厂);薄层板(自制)。
1.2 试药
紫草药材由甘肃省中医院药剂科提供,经甘肃省中医院药学部汪付田主任中药师鉴定为正品;紫草总色素β-环糊精包合物(甘肃省中医院科研制剂中心制备,批号分别为20160906,20160915,20160918,20160920,20160925,20161211);左旋紫草素对照品(中国食品药品检定研究院,批号为110769-200405,供含量测定用);β,β-二甲基丙烯酰阿卡宁对照品(中国食品药品检定研究院,批号为111689-200502);β-环糊精(江苏丰园生物技术有限公司,相对分子质量为1 134.98,含量≥98%);乙腈、甲醇(天津光复精细化工研究所,色谱纯);其他试剂均为分析纯,水为自制超纯水。
2 方法与结果[7]
2.1 薄层色谱鉴别
取总萘醌包合物0.5 g,加石油醚20 mL,超声20 min,滤过,滤液浓缩至约1 mL,作为供试品溶液。同法制得β-环糊精阴性对照品溶液。另取β,β-二甲基丙烯酰阿卡宁对照品、左旋紫草素对照品适量,配成对照品溶液。按2015年版《中国药典(四部)》通则0502薄层色谱法试验,分别吸取供试品溶液、对照品溶液、阴性对照品溶液各10μL,点于同一以羧甲基纤维素钠为黏合剂的硅胶G薄层板上,以环己烷-甲苯-乙酸乙酯-甲酸(5∶5∶0.5∶0.1,V/V/V/V)为展开剂,展开,取出,晾干,喷10%氢氧化钾甲醇溶液,显色。供试品溶液色谱中,在与对照品溶液色谱相应位置上显相同颜色的荧光斑点,阴性对照无干扰(详见图1)。
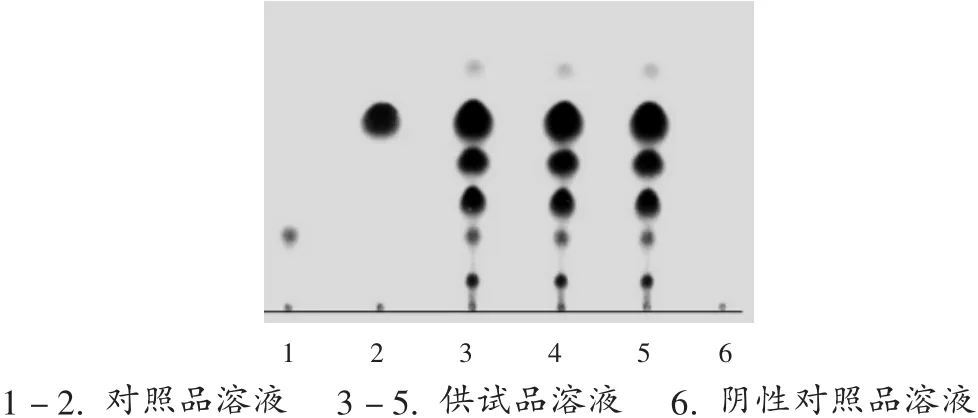
图1 薄层色谱图
2.2 含量测定
2.2.1 色谱条件与系统适用性试验
色谱柱:Waters C18柱(250 mm×4.6 mm,5μm);流动相:甲醇-0.025 mol/L磷酸(82∶18,V/V);流速:0.8 mL/min;检测波长:516 nm;柱温:室温;进样量:10μL[8]。理论板数不低于4 000,且分离度大于1.5。
2.2.2 溶液制备
取左旋紫草素对照品5.11 mg,精密称定,置5 mL容量瓶中,加甲醇配制成质量浓度为1.022 mg/mL的溶液,即得对照品溶液。取包合物0.25 g,置30 mL三角瓶中,加甲醇20 mL,超声(300 W,25 kHz)10 min,0.45μm微孔滤膜过滤,取续滤液,即得供试品溶液。按包合物的处方比例称取环糊精制得阴性样品,按供试品溶液制备方法制得阴性对照品溶液。
2.2.3 方法学考察
专属性试验:分别精密吸取供试品溶液、对照品溶液和阴性对照品溶液,按拟订色谱条件进样测定。结果供试品溶液与对照品溶液在相同时间出峰,左旋紫草素达到较好分离,峰形良好,阴性对照无干扰。色谱图见图2。
线性关系考察:吸取对照品溶液适量,分别用甲醇稀释至每1 mL含左旋紫草素20.4,40.8,81.6,163.2,326.4μg的溶液,按拟订色谱条件进样10μL,每个质量浓度进样3次,以质量浓度(X)为横坐标、峰面积(Y)为纵坐标绘制标准曲线,得回归方程Y=9×105X-50 121,r=0.999 6。结果表明,左旋紫草素质量浓度在20.4~326.4μg/mL范围内与峰面积线性关系良好。
精密度试验:精密吸取质量浓度为81.6μg/mL的对照品溶液10μL,按拟订色谱条件分别于不同时间连续进样6次。结果左旋紫草素峰面积的RSD为0.85%(n=6),表明仪器精密度良好。
稳定性试验:精密吸取同一供试品溶液,分别在0,4,8,12,18,24 h时按拟订色谱条件各进样10μL测定。结果左旋紫草素含量的RSD为1.26%(n=6),表明供试品溶液在24 h内稳定性良好。
重复性试验:取同一批样品(批号为20160925)6份,同法制备供试品溶液,精密吸取10μL进样,记录峰面积。结果样品中左旋紫草素的平均含量为5.58 mg/g,RSD为1.46%(n=6),表明方法重复性良好。
加样回收试验:称取已知含量的包合物(批号为20160906)0.25 g(左旋紫草素含量为5.3 mg/g),精密称定,共6份,分别加入1.022 mg/mL左旋紫草素对照品溶液1 mL,按2.2.2项下样品处理方法处理,按拟订色谱条件进样测定含量,计算回收率。结果见表1。
2.4 样品含量测定
取已制备的3批样品(批号分别为20160906,20160920,20160925),依法制备供试品溶液,按拟订色谱条件平行进样3次,记录峰面积,以外标法计算含量。结果3个批次样品中左旋紫草素含量分别为5.3,5.4,5.6 mg/g。
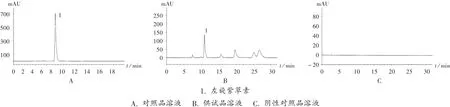
图2 高效液相色谱图
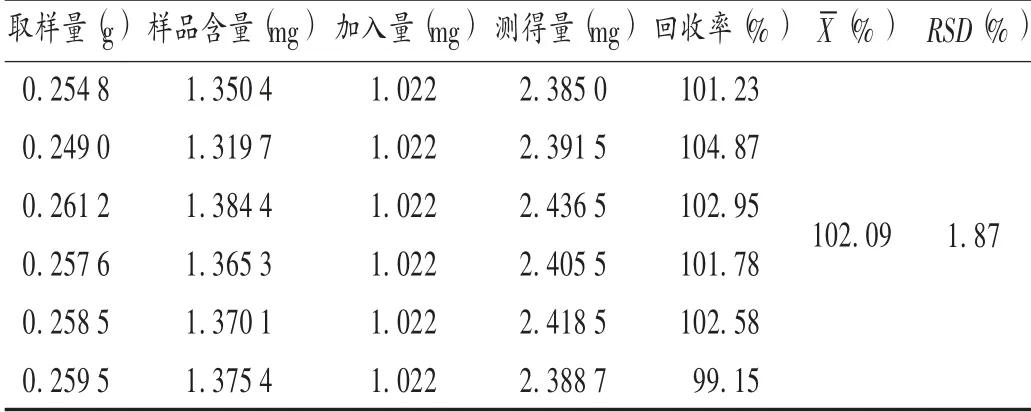
表1 左旋紫草素加样回收试验结果(n=6)
3 讨论
紫草临床应用广泛,主要用于治疗血热毒盛、斑疹紫黑、疮疡、湿疹、水火烫伤、冻伤、流行性腮腺炎、色素性紫癜和过敏性紫癜等,尤其对糖尿病造成的大面积溃疡伤口的愈合效果很好[3-4,9-10]。另外,以紫草为主药治疗皮肤白斑、银屑病、乳腺增生、神经衰弱、精神分裂症等均可获得较好的临床疗效[2]。近年来,又陆续发现紫草具有保肝、抗肿瘤、免疫调节作用,临床可用于急慢性肝炎、绒毛膜上皮癌等的治疗。紫草药理作用及临床应用很多,如抗菌抗炎、抗病毒、抗癌、抗生育、抗氧化、抗衰老、降糖、保肝及提高机体免疫功能[2-4,11-15]。
羟基萘醌化合物为紫草色素的主要组成部分,化学结构由母体萘茜(5,8-dihydroxy-y-1,4-naphthoquinone)与侧链部分相连而成,化学性质与母体有许多相似之处[6]。羟基的引入使得其比母体更不稳定,但具有更丰富的特性。羟基萘醌具有重要生物活性和应用前景,但易受自然界光照、温度、pH、氧化剂、金属离子等的影响而发生降解、氧化、聚合、螯合等变化,改变其稳定性[5-6]。紫草的诸多药理活性使其具有广阔的临床应用前景,但因紫草总色素及衍生物长时间在光、空气和高温环境下产生聚合作用,聚合后则失去活性,口服制剂的稳定性较差,导致紫草的临床应用经常以紫草油等外用剂型多见,单味及复方的口服制剂较少见,临床使用受到了较大限制。
β-环糊精包合物是一种超微型的药物载体,原料是环糊精(CYD)。β-CYD是以7个葡萄糖分子以1,4-糖苷键连接,筒状结构,筒内壁空腔为0.6~1.0 nm,由于葡萄糖的羟基分布在筒的两端并在外部,糖苷键氧原子位于筒的中部并在筒内,β-CYD的两端和外部为亲水性,而筒的内部为疏水性,借范德华力将一些大小和形状合适的药物分子包合于环状结构中,形成超微囊状包合物,供口服或注射,在体内经酶水解释放出药物[16]。采用β-环糊精包合技术克服因有效成分性质不稳定给提取和制备带来的困难,为含紫草的复方及单味药内服制剂的研制提供了创新思路和技术支持,可稳定含紫草的内服中药制剂的质量,提高生物利用度,充分发挥其抗菌抗炎、抗病毒、抗癌、降糖、保肝及提高机体免疫的临床功效。
本研究中对紫草总色素β-环糊精包合物进行定性鉴别和定量测定,采用TLC法、HPLC法对紫草总色素β-环糊精包合物进行质量评价。根据环境和实验室条件,在HPLC-DAD法测定左旋紫草素含量时,发现若单用甲醇水溶液作为流动相,则左旋紫草素峰形会发生拖尾。加入低浓度酸如0.025 mol/L磷酸,能较明显地改善峰形[17]。所建立的TLC定性鉴别及HPLC含量测定方法,完善并提高了紫草总色素β-环糊精包合物的质量控制标准。该方法操作简便、结果准确、专属性强、重复性好,为紫草总色素β-环糊精包合物质量提供了保障,为含紫草的新制剂研发提供了参考。
猜你喜欢
纺织科技进展(2021年3期)2021-06-09
食品研究与开发(2020年13期)2020-07-17
天然产物研究与开发(2020年4期)2020-06-02
分析化学(2019年8期)2019-09-19
中成药(2019年4期)2019-06-01
分析化学(2019年3期)2019-03-30
中国中药杂志(2017年8期)2017-05-11
药学研究(2015年11期)2015-12-19
分析化学(2015年6期)2015-06-18
祝您健康(1995年5期)1995-12-30
