
LaCo1-xCexO3 催化剂在正庚烷催化降解中的性能
2020-02-07 07:00宋一帆徐思遥徐宏泽
化学反应工程与工艺 2020年2期
李 森,宋一帆,徐思遥,刘 虹,徐宏泽
1.上海化工研究院有限公司,上海 200062;
2.上海化工研究院有限公司 聚烯烃催化技术与高性能材料国家重点实验室,上海 200062;
3.上海化工研究院有限公司 上海市聚烯烃催化技术重点实验室,上海 200062
挥发性有机物(VOCs,Volatile Organic Compounds)主要来自于石油化工、制药、印刷、制鞋和喷漆等行业[1]。烷烃、芳香烃、芳烃类、烯烃、醇类、酮类、醛类以及卤代烃等是常见的挥发性有机物[2]。挥发性有机物由于其潜在的高毒性和易在大气中大面积扩散间接形成光化学烟雾等特点而受到特别关注。目前,针对高浓度VOCs 的处理,燃烧法是最方便的措施[3],其中催化燃烧是一种最有效、最经济和最绿色的处理VOCs 的方法。这是因为活性氧参与深度氧化作用后,降低反应的活化能,提高反应速率,并且反应物被分解为二氧化碳和水[4]。催化燃烧法的核心要素之一是催化剂。钙钛矿型催化剂具备良好的热稳定性和耐化学腐蚀性,适合催化燃烧复杂、高浓度的VOCs废气[5]。钙钛矿型催化剂是一类化合物的总称,其化学通式为ABO3,呈立方体晶型。用过渡金属离子或稀土离子部分替代原有位置上的离子,钙钛矿型催化剂仍然保持其立方体晶型,然而稀土离子过量掺杂会使钙钛矿型复合氧化物的晶格发生畸变,甚至钙钛矿晶型被完全破坏[6]。
铈主要以正三价和正四价两种离子的形式存在,具有储存和释放氧的能力[7]。在前期,课题组利用共沉淀法制备了Ce 和La 取代Cu-Co 复合氧化物,发现改性后的Cu-Co 催化剂降解正庚烷的活性显著提高[8-9]。本研究以LaCoO3为前提,通过氨水共沉淀法制备了系列LaCo1-xCexO(3x为0.00,0.05,0.10,0.20,0.30 和0.40)催化剂,并用X 射线衍射(XRD)、多层吸附比表面积测试(BET)、扫描电子显微镜(SEM)、氢气程序升温还原(H2-TPR)和氧气程序升温脱附(O2-TPD)对催化剂进行了表征,探讨了Ce 掺杂量对催化剂的结构、性能以及正庚烷催化燃烧活性的影响。
1 实验部分
1.1 催化剂的制备
钙钛矿LaCo1-xCexO3(x为0.00,0.05,0.10,0.20,0.30 和0.40)催化剂采用共沉淀法制备。按物质的量之比为1:(1-x):x称取La,Co 和Ce 各元素的硝酸盐溶于去离子水中,硝酸盐混合溶液总浓度为0.1 mol/L。用质量分数10%的氨水作为沉淀剂,在室温下,缓慢滴加10%的氨水到搅拌中的硝酸盐混合溶液中,使其pH 值为8.0 在30 min 内保持不变。室温下老化16 h 后抽滤,去离子水洗涤3 次,110 ℃下干燥12 h,然后在马弗炉650 ℃下焙烧6 h 得到钙钛矿催化剂。
1.2 催化剂结构表征
催化剂样品晶型结构在瑞士Thermo Fisher 科技公司的Arlx TRA 型X 射线衍射仪上表征,测试条件为:Cu Kα射线,管电压40 kV,管电流40 mA,扫描速度0.02 (°)/s,扫描范围为5~80°。
比表面积及孔径分布在物理吸附分析仪(V-Sorb 2800TP 型,北京金埃谱科技有限公司)上进行分析,采用Brunauer-Emmett-Teller(BET)方法测定催化材料的比表面积,采用Horvath-Kawazoe(HK)方法计算催化材料的微孔孔径分布。
催化剂的形貌在德国卡尔蔡司公司的Melin Compact 型扫描电子显微镜上进行,样品固定在碳基材料上,放入电镜预处理室,抽取真空后转入测量室,放大20 000 倍,观察样品形貌并拍摄照片。
催化剂的氧化还原性能由氢气程序升温还原(H2-TPR)实验测定,在浙江泛泰仪器有限公司的FINESORB-3010 型全自动程序升温吸附仪上进行,称取50 mg 样品,用石英棉将样品固定于U 型管内,N2氛围下250 ℃预处理0.5 h 后,冷却至室温;切换到氢气/氩气(氢气体积分数为5%)混合气为流动相直到基线稳定,以10 ℃/min 速度程序升温,并使用热导检测器(TCD)检测信号。
催化剂表面活性氧分布情况通过氧气程序升温脱附(O2-TPD)实验测试,在美国麦克仪器公司的AUTOCHEM2920 全自动程序升温吸附仪上进行,测试前,样品(100 mg)在纯O2中400 ℃预处理60 min,冷却至室温后,采用纯He 气净化至基线稳定。以10 ℃/min 升温速率加热至700 ℃。
1.3 催化剂活性评价
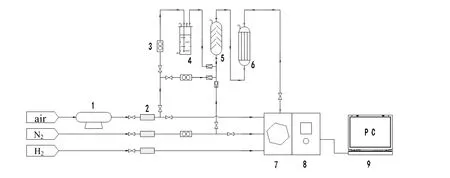
图1 固定床连续反应器-气相色谱在线检测装置 Fig.1 Continuous fixed bed reactor- gas chromatography online inspection apparatus
活性测试在自制固定床连续反应器-气相色谱在线检测装置上进行,见图1。VOCs 催化剂燃烧 性能测试在内径为4 mm 石英反应管内进行,催化剂用量0.15 g,并用0.75 g 石英砂均匀混合。装有正庚烷的鼓泡器置于装有冷媒的杜瓦瓶中,使有机物蒸汽压保持在0 ℃时测得。用氮气稀释正庚烷/空气混合气体,使正庚烷体积分数为0.18%,空速为15 000 h-1。
2 结果与讨论
2.1 XRD 测试
如图2 所示,钙钛矿LaCoO3的特征主峰在2θ为23,33,47 和58°左右,并且分成两个子峰,与标准LaCoO3(P750279)相符。由图2 可知,LaCo1-xCexO3(x为0,0.05 和0.10)样品显示出尖锐而强烈的峰值,表明部分Ce 取代相应位置上的Co 后仍呈现单一的LaCoO3钙钛矿结构。LaCo1-xCexO3(x为0.20,0.30 和0.40)样品除了有钙钛矿状结构外,在28.5 和55.4°处有尖而宽的CeO2的信号峰,随着取代比例的增加,钙钛矿结构受到部分取代破坏,CeO2晶相逐渐增多。这说明并非所有的Ce 都是完全取代LaCoO3晶体中Co 位结构体,Ce 的掺杂导致了LaCo1-xCexO3晶相缺陷的产生,此时催化剂中不是按化学计量形成的LaCo1-xCexO3纯钙钛矿相,而是LaCo1-xCexO3钙钛矿相和CeO2相的混合体系。随着铈的掺杂,钙钛矿的特征峰位置略有移动,峰形变宽,峰强度减弱。在2θ为33°时,有部分峰发生分裂,这也说明对于钙钛矿型催化剂B 位掺杂量达到一定程度后晶体结构将发生改变。当x为0.40 时,钙钛矿晶型的特征峰强度最弱,CeO2的特征峰强度增强,催化剂的晶相以CeO2为主,大部分钙钛矿晶型被破坏。

图2 LaCo1-xCexO3 系列样品催化剂的XRD 图谱 Fig.2 XRD patterns of series of LaCo1-xCexO3 catalysts
2.2 BET 测试和SEM 表征
表1 为LaCo1-xCexO3催化剂比表面积分析结果。由表可看出,不同Ce 掺杂量的LaCo1-xCexO3比表面积在3.9~11.6 m2/g。与LaCoO3催化剂相比,随着Ce 掺杂量的增加,催化剂的比表面积基本呈现增大的趋势。结果表明铈掺杂提高了比表面积。根据XRD 分析结果可以看出,Ce 含量的增加导致催化材料结构中CeO2晶相逐渐增多,尤其当x为0.40 时,钙钛矿晶型被完全破坏,CeO2晶相为催化剂的主要晶型。根据文献[10]可知,相比于钙钛矿,氧化铈比表面积大些。因此,表1 中催化剂的高比表面积是大量氧化铈作用的结果。当掺杂Ce 的比例过大时,催化剂大多以氧化铈晶型为主,并伴有少量钙钛矿晶型。为了保证催化剂既有完整的钙钛矿晶型又有较大的比表面积,Ce替代Co 的量为0.05 时最佳。

表1 LaCo1-xCexO3 催化剂的结构性能 Table 1 Structure characteristics of LaCoxCe1-xO3 catalysts
图3 为LaCo1-xCexO3催化剂的SEM 结果。由图可以看出,LaCoO3催化剂除了存在层状颗粒结构外,还有部分棒状分布,这与文献报道相符,属于钙钛矿颗粒聚集体的特征形貌[11]。Ce 部分掺杂Co 后,催化剂形貌仍然为“颗粒聚集”,但当x为0.40 时,催化剂只有少量的层状颗粒分布,钙钛矿特征形貌被破坏。从表1 和图3 分析可知,Ce 的掺杂不仅会影响催化剂的形貌还会影响催化剂表面结构,从而对催化反应的扩散效应造成影响。

图3 催化剂的SEM 图片 Fig.3 SEM images of catalysts
2.3 H2-TPR 表征
图4 为LaCo1-xCexO3催化剂的H2-TPR 谱图。钙钛矿催化剂的活性主要取决于催化剂中B 位替代离子的氧化形态,活性组分的可还原性与催化氧化的活性直接相关[10]。由文献可知,LaCoO3有2 个还原峰,低温还原峰发生式(1)所示的反应,在这个过程中Co3+转变为Co2+,同时有LaCoO2.5钙铁石的形成[12]。而高温还原峰则发生式(2)所示的反应,Co2+进一步还原为Co0,生成金属钴[12]。

由图4 可以看出,LaCo1-xCexO3(x为0.05,0.10,0.20,0.30 和0.40)催化剂有两个明显的特征还原峰,低温还原峰在250~300 ℃,认为是归属于Co3+的还原,还有1 个在375~475 ℃的高温还原峰,是二价钴还原为Co0产生的还原峰。在800 ℃ 左右,纯的CeO2还原为Ce2O3,但很难还原到Ce0[8],这就是H2-TPR 谱图中未出现CeO2还原峰的原因。LaCo1-xCexO3催化剂的低温还原 峰随着Ce 取代比例的增加而向低温方向发生迁移,这可能与掺杂Ce 后B 位Co 元素形成的Co3+价态向Co2+价态的还原有关[13]。对于掺杂Ce 的催化剂样品,所有低温还原峰都向低温区方向移动,Ce的掺杂使催化剂氧空穴处的化学吸附氧容易移动,提高了催化剂的活性,有利于低温氧化反应,B 位上的金属离子产生强烈的相互作用[14],从而提高了 LaCo1-xCexO3催化剂氧化还原能力。与LaCo1-xCexO3(x为0.00,0.10,0.20,0.30 和0.40)催化剂相比,LaCo0.95Ce0.05O3催化剂的高温还原峰温度略高,这说明LaCo0.95Ce0.05O3催化剂既有较高的催化活性又有较好的稳定性。
2.4 O2-TPD 表征
图5 为LaCo1-xCexO3催化剂的O2-TPD 谱图。一般来说,LaCoO3在低温处会出现一个较宽较弱的脱附峰(α峰),归属于氧空位上的吸附氧脱附,其脱除部分与Co 位金属物理化学性质有关,主要决定于样品表面氧空位数量;在高温处出现较强的脱附峰,一般归属为β氧,属晶格氧,与Co离子氧化还原作用有关[5]。由图5 可以看出,LaCo1-xCexO3催化剂在250 ℃低温处出现脱附峰,对应化学吸附氧(α氧),随着Ce 含量的增加,α峰的面积先增大后减小,这说明催化剂中普通吸附氧和氧空穴处化学吸附氧也是先增加后减少。在800 ℃ 以上高温区有一个较强的氧脱附峰,产生这个信号峰的原因可能是与晶格氧或与氧物种相关的物质占据了由Ce 代替Co 后产生的内部空位有关。B 位掺杂Ce 后,高温脱附峰的面积随掺杂量的增加先增大后减小,表明催化剂中晶格氧数量先增多后减少。一般认为,参与氧化反应的活性氧种倾向于晶格氧,而化学吸附氧主要起传递作用,晶格氧的增加有利于反应的进行[6]。O2-TPD 表征结果中不同催化剂β峰温度相差不大,但高温脱附峰面积从高到低的顺序为LaCo0.95Ce0.05O3,LaCo0.90Ce0.10O3,LaCo0.90Ce0.20O3,LaCoO3,LaCo0.70Ce0.30O3和LaCo0.60Ce0.40O3,说明LaCo0.95Ce0.05O3催化剂中存在较多的活性氧,有利于氧化反应的进行。

图4 LaCo1-xCexO3 催化剂H2-TPR 图谱 Fig.4 H2-TPR pattern for LaCoxCe1-xO3 catalysts

图5 LaCo1-xCexO3 催化剂O2-TPD 图谱 Fig.5 O2-TPD pattern for LaCo1-xCexO3 catalysts

图6 LaCo1-xCexO3 催化剂在正庚烷燃烧中的催化活性 Fig.6 Catalytic performance of LaCo1-xCexO3 catalysts in n-heptane combustion
2.5 催化降解正庚烷性能
图6 为所制备系列催化剂对正庚烷催化降解的实验结果。由图6 可知,LaCoO3催化剂在降解正庚烷的实验中,对正庚烷转化率达到90%时反应温度需要350 ℃。相对的是,掺杂Ce 的催化剂上正庚烷催化降解的活性均有不同程度的提高,特别是掺杂少量Ce 的催化剂上这一现象最明显,当使用LaCo0.95Ce0.05O3和LaCo0.90Ce0.10O3催化剂时,正庚烷转化率达到90%时的温度分别降至为200 和260 ℃。对LaCo0.95Ce0.05O3催化剂而言,当反应温度为245 ℃时,该催化剂对正庚烷转化率可达99%。上述结果表明,Ce 掺杂取代了钙钛矿中Co,破坏了原有的化学计量平衡,致使催化剂上的氧空位和缺陷增加,与此同时,Ce 和Co 二者的协同效应也使催化剂活性明显提高[15]。但是当Ce 含量进一步增加,即x为0.30 和0.40 时,催化性能没有提高,反而逐渐降低,这主要是由于掺杂离子过多使催化剂结构发生了改变。结合图2 和表1 可知,过多Ce 的添加导致钙钛矿晶型被破坏,虽然催化剂的比表面积增大,但催化剂燃烧活性降低,由此可知,比表面积大小并不是决定催化剂活性的关键因素。关联H2-TPR 及O2-TPD 表征结果可知,LaCo0.95Ce0.05O3和LaCo0.90Ce0.10O3催化剂的高活性表现为其氧空穴处的化学吸附氧容易移动,并且催化剂中晶格氧数量较多,这是由于两个催化剂均保持钙钛矿结构,有利于氧空穴快速和完全的再填充。
3 结 论
a)采用共沉淀法合成出了Ce 掺杂的LaCo1-xCexO3钙钛矿型催化剂,相比于LaCoO3催化剂,当x小于等于0.10 时,催化剂具有单一的钙钛矿晶型;当x大于0.10 时,钙钛矿晶型被破坏,并出现CeO2晶相。
b)当Ce 的添加量为0.05~0.10,正庚烷转化率达到90%时所需反应温度为260 ℃左右,与LaCoO3催化剂相比,反应温度降低近90 ℃。关联表征结果表明,LaCo0.95Ce0.05O3及LaCo0.90Ce0.10O3催化剂既在氧空穴处具有较多的易移动的化学吸附氧,又具有较多的晶格氧,因此提高了催化剂的氧化还原能力。
猜你喜欢
科学技术与工程(2022年26期)2022-11-01
太原理工大学学报(2022年5期)2022-09-23
工程技术与管理(2022年7期)2022-03-04
当代作家(2021年11期)2021-12-17
弹性体(2021年6期)2021-02-12
哈尔滨工业大学学报(2020年1期)2020-12-21
科学(2020年4期)2020-11-26
陶瓷学报(2020年2期)2020-10-27
生物化工(2020年4期)2020-08-27
科学(2020年4期)2020-01-11
