
熊果酸/甘草次酸-尿嘧啶核苷缀合物的合成与抗肿瘤活性评价
2020-01-15 05:12:20初相伍刘春梅张琚政程克光
广西师范大学学报(自然科学版) 2020年1期
孙 立,初相伍,刘春梅,张琚政,程克光*
(1.省部共建药用资源化学与药物分子工程国家重点实验室(广西师范大学),广西 桂林 541004;2.广西师范大学 化学与药学学院,广西 桂林 541004)
肿瘤疾病的发病率不断上升,其死亡率仅次于心血管疾病[1]。手术和化疗仍是主要的有效治疗手段,但具有预后性差和毒副作用大的缺点,因此开发新型的具有靶向性的抗肿瘤候选药物是当今抗肿瘤药物研究的热点[2-3]。将具有抗肿瘤活性的药效团与某种具有靶向性的物质结合获得新的抗肿瘤药物是目前研究者惯用的一种手段[4]。
核苷与大多数生物的生长、遗传和变异息息相关,如核酸和蛋白质的合成、酶的调控、新陈代谢等。近年来,核苷及其衍生物在抗病毒、抗肿瘤等方面得到了广泛的发展[5-6],研究发现尿嘧啶核苷衍生物可以有效作为半乳糖转移酶抑制剂;5′-酰胺基尿嘧啶核苷衍生物是三磷酸核苷二磷酸水解酶-2(NTPDase2)的抑制剂[7-8]。一些核苷类似物已被应用于临床,如5-氟尿嘧啶(5-FU)、氟铁龙(doxifluridine)、卡培他滨(capecitabine)、吉西他滨(gemcitabine)等。
近30年来,有近一半市售的新药是天然产物或天然产物的衍生物[9-10]。五环三萜类化合物在自然界中分布广泛,是很多常用中草药的有效成分,具有广泛的生物活性,如降血糖血脂、抗炎、抗肿瘤、抗HIV、抗病毒等[11],还具有调节耐药性和增加化疗药物敏感性的功能[12]。因此,以五环三萜作为抗肿瘤药物的先导化合物的研究越来越多。熊果酸(UA)和甘草次酸(GA)都是典型的五环三萜化合物。研究发现,用聚乙二醇修饰的熊果酸衍生物增强了熊果酸的水溶性并且提高了它的体外抗癌活性[13],熊果酸A环衍生物和羧酸酯化衍生物对Hep G2、SGC7901肿瘤细胞的抑制作用均强于母体熊果酸[14]。熊果酸对人慢性髓原白血病细胞(K562)通过增加Caspase-3的活性,上调凋亡促进因子Bax的表达,下调BCR/ABL中的酪氨酸酶的活性,从而诱导细胞凋亡[15]。甘草次酸的C3位羟基、C30位羧基经结构修饰的衍生物对MCF-7和A549癌细胞表现出明显高于对照药吉非替尼的抑制活性[16]。
本课题组前期研究显示,齐墩果酸-尿嘧啶核苷缀合物具有较好的抗肿瘤活性,活性最好的化合物其IC50低于0.1 μmol/L[4]。为了探索其他五环三萜与尿嘧啶核苷缀合后的抗肿瘤活性,本文以具有抗肿瘤活性的天然产物熊果酸和甘草次酸为母体,设计、合成了4个熊果酸/甘草次酸-尿嘧啶核苷缀合物,并对它们进行了初步的体外抗肿瘤活性研究。如图1所示,分别以熊果酸和甘草次酸为原料,在K2CO3碱性条件下,经二溴烷烃酯化,获得溴代烷烃酯化合物1a~4a[17-18];然后与尿嘧啶核苷发生亲核取代反应,得熊果酸/甘草次酸-尿嘧啶核苷衍生物1b~4b。采用MTT法[19]对合成的目标物1b~4b进行体外抗肿瘤活性测试。
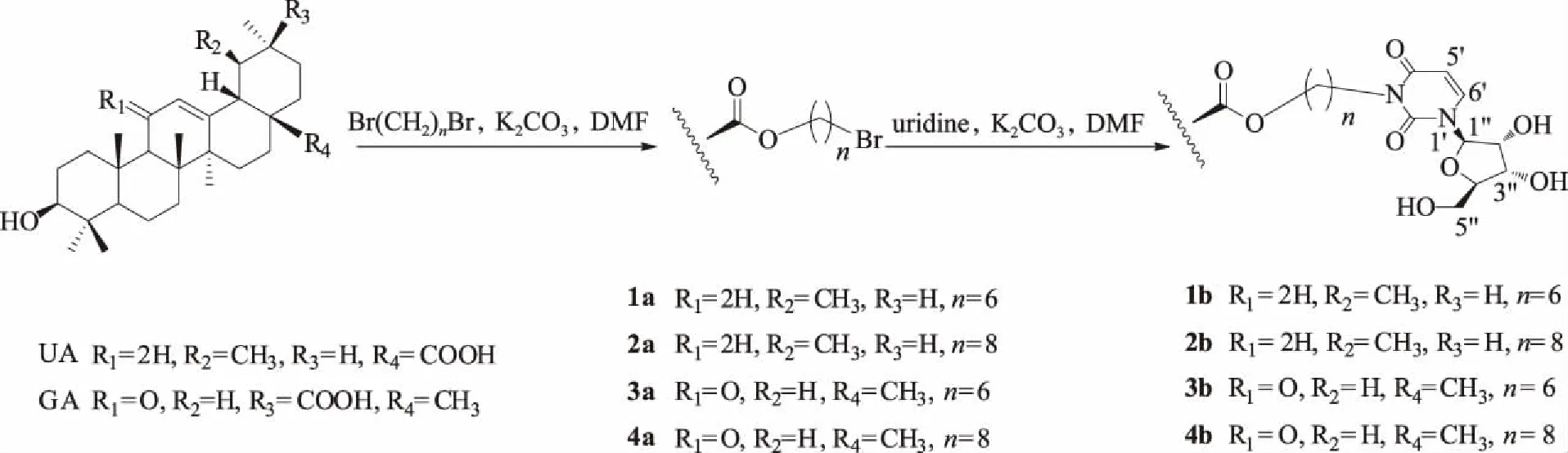
n表示亚甲基的数目图1 熊果酸/甘草次酸-尿嘧啶核苷缀合物的合成Fig.1 Synthesis of uridine-UA/GA conjugates
1 实验部分
1.1 仪器与试剂
Bruker AV-500和AV-300 超导核磁共振仪、Esquire HCT 型离子阱质谱仪、HCT 电喷雾质谱(美国Bruker 公司);RY-1熔点测定仪(天津天分分析仪器厂);DLSB-5/20型低温冷却液循环泵(郑州长城科工贸有限公司);AG 22331 Hamburg离心机(德国Eppendorf公司);LX51显微镜(日本奥林巴斯公司);M1000酶标仪(瑞士帝肯集团公司)。
所有化学合成用原料均为化学纯或分析纯试剂;胰蛋白酶消化液、RPMI-1640、胎牛血清、DMSO、PBS等均购于阿法埃莎(天津)化学有限公司。
1.2 实验方法
1.2.1 化合物1a~4a的制备
将熊果酸(0.50 g,1.09 mmol)溶于DMF(5 mL)中,加入K2CO3(0.15 g, 1.09 mmol)和1,6-二溴己烷(0.84 mL, 5.47 mmol),30 ℃下搅拌反应12 h。浓缩反应液,残余物分散在乙酸乙酯(50 mL)中,依次用1 mol/L HCl、饱和NaHCO3和饱和食盐水洗涤,无水Na2SO4干燥,过滤,滤液蒸除溶剂得粗产物。硅胶柱层析分离(petroleum ether∶EtOAc=3∶1,体积比,下同)得化合物1a[17-18](0.61 g, 产率90.0%, 白色固体)。
将熊果酸(2.00 g, 4.38 mmol)和1,8-二溴辛烷(2.40 mL, 13.14 mmol)同法操作,硅胶柱层析分离(petroleum ether∶EtOAc=4∶1),得化合物2a[17-18](2.30 g, 产率81.0%, 白色固体)。
将甘草次酸(2.00 g, 4.25 mmol)和1,6-二溴己烷(3.24 mL, 21.25 mmol)同法操作,硅胶柱层析分离(petroleum ether∶EtOAc=5∶2),得化合物3a[18,20](2.37 g, 产率88.0%, 白色固体)。
将甘草次酸(2.00 g, 4.25 mmol)和1,8-二溴辛烷(3.24 mL, 21.25 mmol)同法操作,硅胶柱层析分离(petroleum ether∶EtOAc=5∶2),得化合物4a[18,20](2.42 g, 产率86.0%, 白色固体)。
1.2.2 化合物1b~4b的制备
将化合物1a(0.40 g, 0.65 mmol)溶于DMF(6 mL)中,加入K2CO3(0.27 g, 1.94 mmol)和尿嘧啶核苷(0.47 g, 1.94 mmol),50 ℃下搅拌反应过夜。减压旋除溶剂,残余物用水(50 mL)分散,乙酸乙酯(20 mL×3)萃取,合并有机层,依次用HCl、饱和NaHCO3、饱和食盐水洗涤,无水Na2SO4干燥,过滤,滤液浓缩,所得残留物经硅胶柱层析分离纯化(petroleum ether∶EtOAc=1∶10)得化合物1b(0.26 g, 产率52.8%,白色固体)。m.p.120~122 ℃。1H NMR (300 MHz, CDCl3):0.74, 0.78, 0.85 (d, 3H,J=6.4 Hz, CH3), 0.92 (d, 3H,J=3.8 Hz, CH3), 0.95, 0.99, 1.07 (5 s, each 3H, 5×CH3), 0.79~2.10 (m, 30H), 2.20 (d, 1H,J=11.1 Hz, H-18), 3.22 (dd, 1H,J=5.0, 10.7 Hz, H-3), 3.80~4.43 (m, 10H, H-2″, H-3″, H-4″, H-5″, OH, COOCH2, NCH2), 5.23 (s, 1H, H-12), 5.63 (d, 1H,J=4.3 Hz, NCHO), 5.76 (d, 1H,J=8.1 Hz, CCH=), 7.60 (d, 1H,J=8.1 Hz, NCH=)。13C NMR (75 MHz, CDCl3)15.3, 15.6, 16.9, 17.0, 18.2, 21.1, 23.2, 23.4, 24.1, 25.5, 26.3, 27.0, 27.3, 27.9, 28.1, 28.2, 30.6, 32.9, 36.7, 36.9, 38.5, 38.6, 38.8, 39.0, 39.5, 40.9, 42.0, 47.4, 48.0, 52.8, 55.1, 61.7, 64.0, 70.3, 74.8, 79.0, 85.4, 93.3, 101.8, 125.5, 138.1, 138.8, 151.5, 162.5, 177.9。HRMS (ESI)(m/z):[M+Na]+calcd for C45H70N2O9Na 805.497 9, found 805.498 6。
将化合物2a(0.40 g, 0.63 mmol)和尿嘧啶核苷(0.46 g, 1.89 mmol)同法操作,硅胶柱层析分离(petroleum ether∶EtOAc=1∶10),纯化得化合物2b(0.16 g, 产率31.6%, 白色固体)。m.p. 106~108 ℃。1H NMR (300 MHz, CDCl3): 0.74, 0.78, 0.85 (d, 3H,J=6.5 Hz, CH3), 0.92, 0.94 (d, 3H,J=6.3 Hz, CH3), 0.98, 1.07 (5 s, each 3H, 5×CH3), 0.79~2.10 (m, 34H), 2.22 (d, 1H,J=12.1 Hz, H-18), 2.54 (s, 1H, OH), 3.22 (dd, 1H,J=10.4, 5.4 Hz, H-3), 3.32 (s, 1H, OH), 3.81~4.37 (m, 10H, H-2″, H-3″, H-4″, H-5″, OH, COOCH2, NCH2), 5.23 (s, 1H, H-12), 5.63 (d,J=3.6 Hz, 1H, NCHO), 5.75 (d, 1H,J=8.1 Hz, CCH=), 7.60 (d, 1H,J=8.1 Hz, NCH=)。HRMS (ESI) (m/z):[M+Cl]-calcd for C47H74N2O9Cl 845.508 3, found 845.509 3。
将化合物3a(0.40 g, 0.63 mmol)和尿嘧啶核苷(0.46 mg, 1.90 mmol)同法操作,硅胶柱层析分离(petroleum ether∶EtOAc = 1∶10),纯化得化合物3b(0.23 g, 产率46.4%, 白色固体)。m.p.120~122 ℃。1H NMR (300 MHz,CDCl3):0.80, 1.00, 1.12 (s, 6H, 2×CH3), 1.13, 1.14, 1.37 (5 s, each 3H, 5×CH3), 0.67~2.34 (m, 27H), 2.35 (s, 1H, H-9), 2.75 (d, 1H,J=13.5 Hz, H-18), 3.23 (s, 1H, OH), 3.36 (s, 1H, OH), 3.45 (d, 1H,J=3.4 Hz, H-3), 3.82~4.60 (m, 10H, H-2″, H-3″, H-4″, H-5″, OH, COOCH2, NCH2), 5.63 (s, 1H, H-12), 5.72 (d, 1H,J=8.1 Hz, CCH=), 5.79 (d, 1H,J=4.7 Hz, NCHO), 7.77 (d, 1H,J=8.1 Hz, NCH=)。HRMS (ESI) (m/z): [M+Na]+calcd for C45H68N2O10Na 819.477 2, found 819.479 9。
将化合物4a(0.40 g, 0.62 mmol)和尿嘧啶核苷(0.45 mg, 1.85 mmol)同法操作,硅胶柱层析分离(petroleum ether∶EtOAc=1∶10),纯化得化合物4b(0.12 g, 产率45.3%, 白色固体)。m.p.104~106 ℃。1H NMR (300 MHz, CDCl3): 0.80, 1.00, 1.14, 1.33, 1.36 (5 s, each 3H, 5×CH3), 1.12 (s, 6H, 2×CH3), 0.64~2.35 (m, 31H), 2.35 (s, 1H, H-9), 2.73 (d, 1H,J=13.4 Hz, H-18), 3.01 (s, 1H, OH), 3.22 (d, 1H,J= 3.0 Hz, H-3), 3.50 (s, 1H, OH), 3.81~4.38 (m, 10H, H-2″, H-3″, H-4″, H-5″, OH, COOCH2, NCH2), 5.64 (s, 1H, H-12), 5.75 (d, 1H,J=8.1 Hz, CCH=), 5.71 (d, 1H,J=3.0 Hz, NCHO), 7.69(d, 1H,J=8.1 Hz, NCH=)。HRMS (ESI) (m/z): [M+Na]+calcd for C47H72N2O10Na 847.508 5, found 847.509 5。
1.3 化合物的抗肿瘤活性测试
以相应溶剂作空白对照,5-氟尿嘧啶为阳性药物, HepG2(人肝癌细胞株)、A549(人肺癌细胞株)、HeLa(人宫颈癌细胞株)、BGC-823(人胃腺癌细胞株)、NCI-H460(人肺癌细胞株)、BEL-7404(人肝癌细胞株)、HL-7702(人正常肝细胞)等7株细胞为测试细胞株,用MTT 法对目标化合物进行体外抗肿瘤活性测试。所有细胞株均培养在含有10%胎牛血清的DMEM培养基中,在37 ℃、 CO2体积分数5%的细胞培养箱中培养至细胞对数生长期。将待测化合物以DMSO助溶后,使用PBS缓冲液配成1 000、500、100、50、10和1 μmol·L-1的工作液,存放于4 ℃冰箱保存,供测试受试化合物对所选肿瘤细胞株的IC50值使用。
取处于对数生长期状态良好的细胞一瓶,加入质量分数0.25%胰蛋白酶消化液,消化使贴壁细胞脱落,计数2×104~4×104个/mL,制成细胞悬液;取细胞悬液接种于96孔板上,180 μL/孔,置恒温CO2培养箱中培养24 h;换液,加入受试化合物,20 μL/孔,培养72 h;将MTT加入96孔板中,20 μL/孔,培养箱中反应4 h;吸去上清液,加入DMSO,100 μL/孔,平板摇床上振摇10 min;用酶联免疫检测仪在波长为570 nm处测定每孔的吸光值,并计算细胞抑制率(细胞抑制率=(阴性对照组OD值-受试药物组OD值)/阴性对照组OD值×100%),测得各受试化合物的体外抗肿瘤细胞增殖活性数据。表1为20 μmol/L测试化合物对不同肿瘤细胞株的抑制率及其IC50。
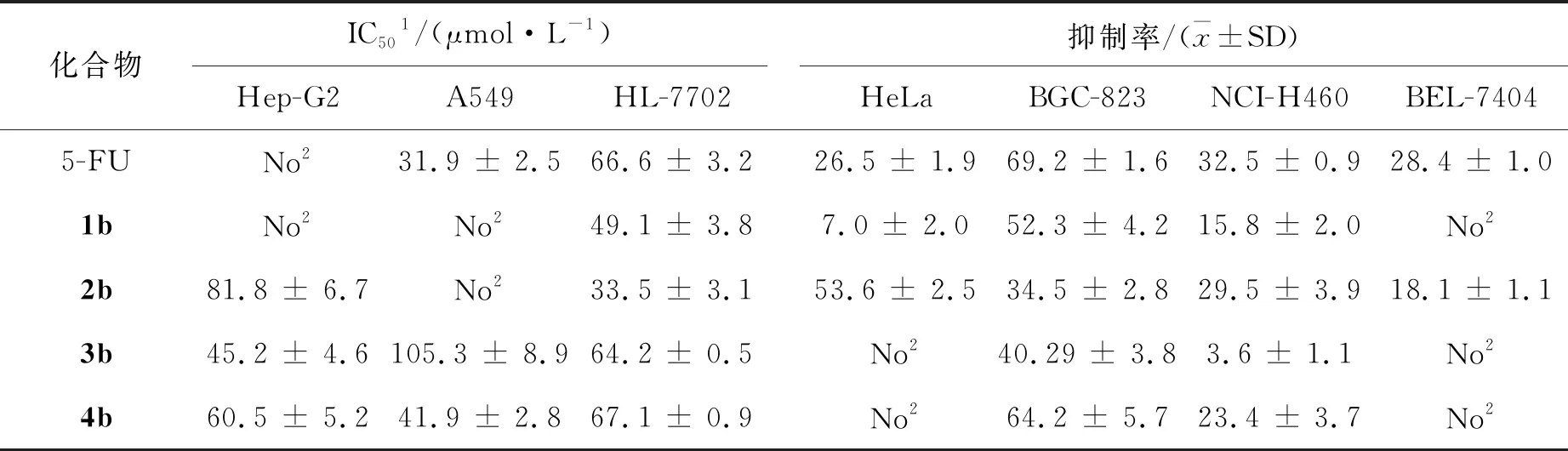
表1 测试化合物对不同肿瘤细胞株的IC50 和抑制率
2 结果分析与讨论
从表1数据可知,甘草次酸-尿嘧啶核苷类缀合物3b和4b对HepG-2和A549的抑制作用强于熊果酸-尿嘧啶核苷类缀合物1b和2b。熊果酸-尿嘧啶核苷类缀合物中,化合物2b对Hep-G2的抑制活性相对强一些,但同时毒性较强,对正常细胞HL-7702具有明显抑制作用。甘草次酸-尿嘧啶核苷类衍生物3b和4b对HepG-2具有一定的抑制作用,对正常细胞HL-7702也有一定毒性。熊果酸/甘草次酸-尿嘧啶核苷类化合物对BGC-823表现出较好的抑制作用,其中4b的活性最好,抑制率达到64.2%。此外,化合物2b对HeLa的作用较好,抑制率达到53.6%。
3 结论
因前期研究表明齐墩果酸-尿嘧啶核苷缀合物具有较好的抗肿瘤活性[4],本文进一步合成了4个熊果酸/甘草次酸-尿嘧啶核苷缀合物,并对它们进行了体外抗肿瘤活性测试。活性测试结果表明,熊果酸/甘草次酸-尿嘧啶核苷衍生物具有一定的抗肿瘤活性,但远不如前期研究工作中的齐墩果酸-尿嘧啶核苷衍生物的抗肿瘤活性。
猜你喜欢
寻根(2022年2期)2022-04-17 11:01:38
核农学报(2020年2期)2020-03-11 08:33:16
中成药(2018年2期)2018-05-09 07:20:08
中成药(2017年3期)2017-05-17 06:08:48
菏泽学院学报(2017年2期)2017-05-16 08:59:11
浙江大学学报(人文社会科学版)预印本(2016年4期)2016-02-28 12:13:47
浙江大学学报(人文社会科学版)预印本(2016年4期)2016-02-28 12:13:45
浙江大学学报(人文社会科学版)预印本(2016年4期)2016-02-28 12:13:44
陕西师范大学学报(自然科学版)(2015年1期)2016-01-16 03:23:30
山东医药(2015年16期)2016-01-12 00:40:04
