
尿嘧啶及其衍生物溶解焓的影响因素探讨
2017-05-16 08:59:11吕惠萍
菏泽学院学报 2017年2期
吕惠萍,朱 琪
(菏泽学院化学化工系,山东 菏泽 274015)
尿嘧啶及其衍生物溶解焓的影响因素探讨
吕惠萍,朱 琪
(菏泽学院化学化工系,山东 菏泽 274015)
在298.15K、一个大气压下,用密度泛函数在B3LYP/6-31+G*水平上利用PCM(Polarizable continuum model)极化连续模型模拟尿嘧啶,5-氟-尿嘧啶,5-氯-尿嘧啶,5-溴-尿嘧啶和5-三氟-尿嘧啶溶入甲醇和水中的焓值.计算所得尿嘧啶及其卤素衍生物在水中的溶解焓与实验值一致,但尿嘧啶及其卤素衍生物在甲醇中的溶解焓与实验值差别较大.由此分析了分子在两种溶剂中的氢键、电子密度拉普拉斯值以及它们的π键的影响,结果表明溶于甲醇溶液的氢键键能偏大,电子密度拉普拉斯值以及分子的π键与实验结果有良好的相关性.
尿嘧啶;尿嘧啶衍生物;溶解焓;氢键;电子密度拉普拉斯值;π键
引言
核酸作为重要的生命物质,是遗传信息的携带者和基因表达的物质基础保障.随着现代分子生物学与生物技术的发展,关于核酸的研究以及核酸代谢产物与各类物质相互作用的化学机理研究已经成为核酸研究的重要组成部分[1].尿嘧啶及其衍生物作为医药中间体能够合成尿嘧啶替片,可以合成抗癌药物的中间体广泛地应用于医药科学研究领域,对治疗肠癌、乳腺癌等癌症有很好的临床效果[2].研究此类化合物的热力学特性不但可以促进相应合成工艺的研究[3],而且还能为药物的反应机理和分子结构与性质的相关研究提供宝贵的实验数据和理论依据[4].由于生物体内含有大量的水[5],因此作为基因载体之一的尿嘧啶在水中的溶解是一项值得研究的课题.为了了解结构对溶解焓的影响,我们研究了一系列尿嘧啶及其环衍生物的溶解焓、氢键键长、π键的能量;同时为了了解溶剂的作用,又研究了上述分子在极性较小的甲醇中的溶解焓、氢键键长、以及π键能量的高低.本文通过量子化学的密度泛函理论计算溶质-溶剂相互作用的焓,并从所研究分子形成的氢键键长分析它们结合能的大小,从离域化的π键形式和能量变化分析不同极性的溶剂对溶剂化所起的作用.
1 计算方法
(1)
量化模拟则是计算1 mol溶质溶于大量的水和甲醇溶剂中来获得标准摩尔焓值,同时计算溶质分子在气相中的稳定结构来获得焓值,前者与后者的差值即为溶质分子的标准摩尔溶解焓,即分子从气相溶解到溶液中.计算应用了PCM(polarizable continuum model)模型,应用密度泛函B3lyp结合6-31+g*基组优化了尿嘧啶及其4个卤素衍生物的在气相、水中、甲醇中的结构,以没有虚频判断为能量的稳定值[6~8].由于氢键的作用不能忽略,因此,计算中同时优化了尿嘧啶及其4个卤素衍生物与H2O分子、CH3OH分子结合的聚合物分别以H2O、CH3OH为溶剂的氢键结构.计算得到的所有物质的焓值以尿嘧啶为基准.所有的计算通过Gaussian 09[9]软件包完成.
2 尿嘧啶及其衍生物的溶解焓
通过计算,获得了尿嘧啶及其4个卤素取代物得到溶解焓,并与实验通过量热计获得的数据进行比较(量热计测量的数据为吸热值,实际为溶质溶解时放热所致,故从溶质的角度分析,应是能量的减少值,所以在实验数值前加了负号).所有数据见于表1和表2.

表1 在B3LYP/6-31+G*水平计算得到的尿嘧啶及其衍生物在水溶液的溶解焓(ΔHwater)及与实验数据对比 kJ/moL

表2 在B3LYP/6-31+G*水平计算得到的尿嘧啶及其衍生物在甲醇溶剂中的溶解焓(ΔHCH3OH)及与实验数据对比 kJ/moL
计算取代原子分别为5-F,5-Cl,5-Br和5-3F尿嘧啶的溶剂化焓值在水中分别为:-24.83,-24.46,-25.58 , -25.32 kJ/mol,计算所显示的能量值与实验测定的数值基本相符,计算方法虽然简单,但结果是可靠的.然而取代原子分别为5-F,5-Cl,5-Br和5-3F尿嘧啶的溶剂化焓值在甲醇中计算的数值与实验数据差距较大,分别为:-21.83,-21.48, -23.04, -21.07 kJ/mol.
3 尿嘧啶及其衍生物的电子密度拉普拉斯值的分析
电子密度拉普拉斯值代表了电子密度在某处的总曲率.若某处曲率为正,代表此处的电子密度是发散的;如果为负,代表此处的电子密度为聚集的[10~11].
(2)
AIM(atomic in molecules)理论中的临界点BCP(bond critical point)一般出现在相邻的互相作用的原子之间,这个点被认为是描述相应两个原子相互作用的关键点,这个点的性质(密度、能量密度、动能密度、源函数等)常被用来讨论成键特征.通过AIM理论可以判断两个原子间相互作用类型,也就是这两个原子间的临界点BCP的电子密度拉普拉斯值的符号.如果符号为负,就被认为是共价作用,其原因是共价相互作用的原子间由于共享电子对儿,会造成电子密度在成键区域聚集;如果符号为正,就被认为是闭壳层相互作用,比如离子键、氢键、卤键、二氢键π-π堆积之类,这些相互作用本质是静电或范德华作用,不是靠共享电子对儿实现的,因此,成键区域没有电子密度的聚集,BCP处拉普拉斯值被顺着键径的方向上的电子密度的正曲率所主导而成为正值.
图1是5-Br-U结合4个甲醇分子的电子密度拉普拉斯值等值线图.图中,浅色代表核临界点(NCP: nucleic critical point),深色代表BCP.实线和虚线分别代表拉普拉斯值为正和为负的区域,该图很清晰地表示出电子密度在哪里聚集.
从图1可以看出,甲醇分子自身的O-H以及C-H、N-H、C-Br之间BCP点落在虚线范围内,因此BCP处拉普拉斯值为负值,正确的体现了这几种键都是共价键.而尿嘧啶分子中的氧原子与甲醇分子中的氢原子,即O-H键之间的BCP点落在实线上也就是正值区,由此推断其为氢键,且尿嘧啶分子中的氢原子与甲醇分子中的氧原子,即O-H键之间的BCP点也落在正值区,由此推断其也为氢键.
从图2可以看出,部分C-H以及 N-H、C-O之间BCP点落在虚线范围内,因此BCP处拉普拉斯值为负值.由于氧和氢的电负性相差不大,所以H-O键是极性共价键,BCP依然是落在负值区域.相比于C-H键,BCP的位置更偏向于H,这是由于氧原子吸电子的能力大于碳原子吸电子的能力,所以,在分子环境中它能够在更大的范围内有更广的密度分布,故O-H键上密度最小的点就是BCP点.

图2 5-F尿嘧啶结合4个甲醇分子的电子密度拉普拉斯值等值线图
图3中a图和b图分别是5-溴-尿嘧啶结合4个甲醇分子以及5-溴-尿嘧啶结合4个水分子的电子密度拉普拉斯值等值线图(其它结构见支持信息,图1).对比两图可以发现,两图的纵坐标起点不同,即5-Br-尿嘧啶结合4个水分子的能量比5-Br-尿嘧啶结合4个甲醇分子降低的多.这是因为水的介电常数高于甲醇的介电常数,所以5-溴-尿嘧啶在水溶液中能量降低得更多,结构更稳定.从拉普拉斯等值图上也可以看出,5-溴-尿嘧啶能量降低的延伸值(单位长度:波尔)在水中比在甲醇中长大约4个波尔.

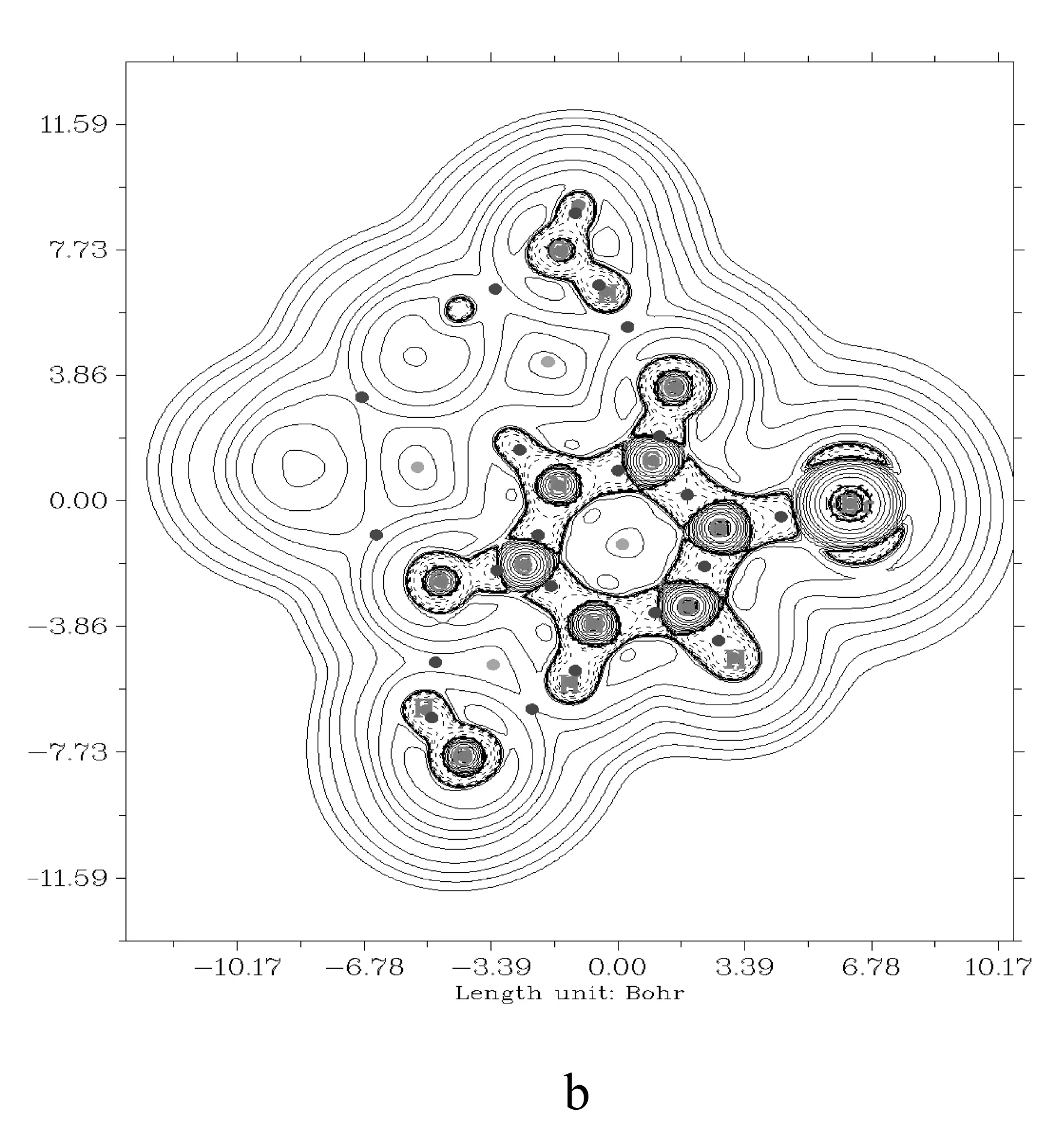
图3 5-溴-尿嘧啶的拉普拉斯等值面,a为甲醇溶剂,b为水溶剂
4 尿嘧啶及其衍生物的π键
π键—原子轨道中两个相互平行的轨道以“肩并肩”方式进行重叠,轨道重叠的部分垂直与键轴并呈镜面反对称分布(原子轨道在镜面两边波的瓣符号相反).
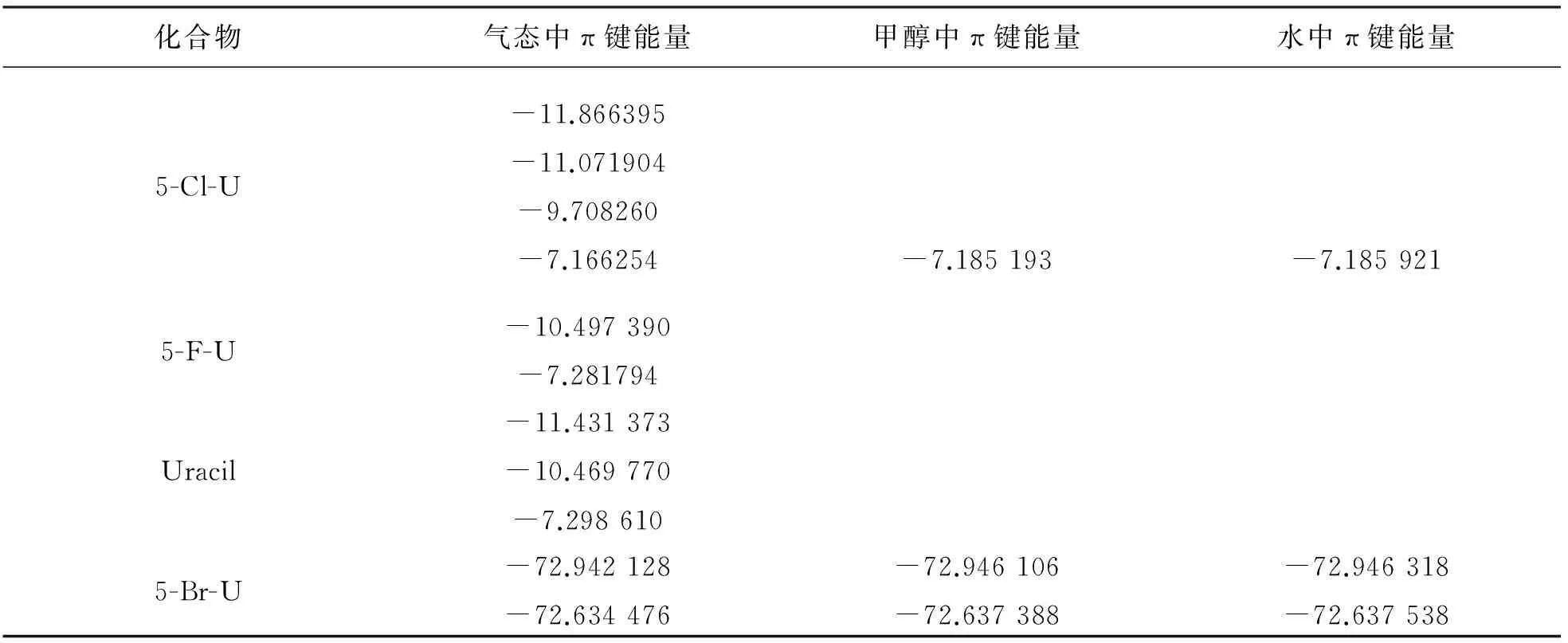
表3 尿嘧啶及其衍生物在气态、甲醇以及水中的π键能量 eV
表3是尿嘧啶及其衍生物的π键分析,是能量变化值.比如5-Cl-U一行,气态下的5-Cl-U共有4个π键,其能量分别为-11.866 395,-11.071 904,-9.708 260,-7.166 254 eV;而结合了甲醇的5-Cl-U的π键能量为-7.185 193 eV,结合了水的5-Cl-U的π键能量为-7.185 192 eV.具体结构如图4所示.
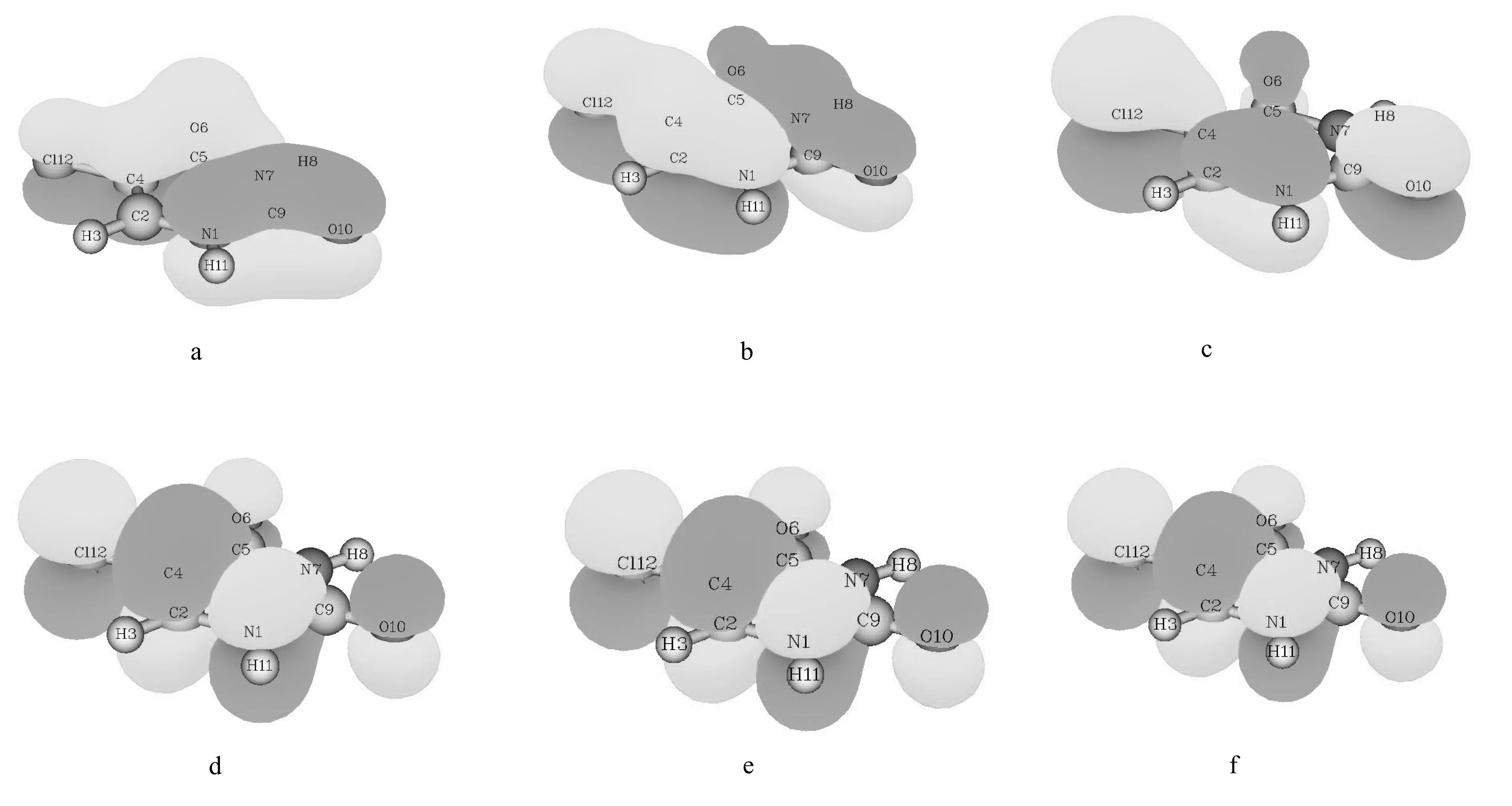
图4 a图,b图,c图,d图是其在气态下的π键图,e图是其溶于甲醇溶液的π键图,f图是其溶于水的π键图.
其中浅色部分是正电荷,而深色部分代表负电荷
由表3中还可以看出,气态下的尿嘧啶及其衍生物的π键最多,而由于溶剂化效应,结合了水和甲醇的π键明显减少甚至没有,并且其π键的能量也明显降低了.以-7.189 519 3 eV为界 ,能量小于此值的π键在结合了水和甲醇后消失.
但5-Br-尿嘧啶例外,气态下的两个π键能量分别为-72.942 128 eV、-72.634 476 eV,小于-7.189 519 3 eV,而结合了水或甲醇后仍然还有两个π键,与上述不符.图5分别是5-Br-尿嘧啶和5-F-尿嘧啶的π键图 (其它结构见支持信息,见图3).从图5中可以看出,两者形成π键的成键位置不同.后者在嘧啶环上形成的是大π键,而前者是在Br上成键,这是因为溶解焓受嘧啶环的影响很大,环外结构较为松散.而溴原子半径大,对电子的排挤性比较大,核外电子多在溴原子上形成π键,所以造成了与上述不符的现象.

图5 a图为5-Br-尿嘧啶的气态π键图,b图为5-F-尿嘧啶的气态π键图
5 总结
研究主要运用量化计算研究了尿嘧啶及其尿嘧啶卤素衍生物在甲醇和水中的溶解焓,并与实验值做了比较.发现,尿嘧啶及其衍生物在甲醇中的溶解焓降低值比在水中的降低值小.从研究可以看出溶剂极性的不同是造成尿嘧啶及其尿嘧啶卤素衍生物溶解焓大小的主要原因.并且解释了尿嘧啶及其尿嘧啶卤素衍生物在甲醇中的溶解焓的计算值远远低于实验值的原因,这是因为PCM溶剂化模型没有考虑微观效应.还列出了5-溴-尿嘧啶溶于甲醇和水中的结构,由于水的体积小,能形成水的链状结构和多个与尿嘧啶结合的氢键,明显看出这有利于降低尿嘧啶(衍生物)的溶解焓.然后又从尿嘧啶(衍生物)的电子密度拉普拉斯值与尿嘧啶及其衍生物的π键分析这两个方面证明上述结论.
[1]张敏,赵虎,张苗,等. 尿嘧啶的荧光猝灭法测定与机理研究[J]. 分析测试学报,2008,08:859-865.
[2]张晓晔,薛斌,程泽,等. 尿嘧啶和5-溴尿嘧啶的低温热容[J]. 物理化学学报,2015,03:412-41.
[3]祁艳. 氟尿嘧啶衍生物及其铜、锰配合物的制备与生物活性研究[D].苏州:江苏大学,2013.
[4]王卫东. 5-氟尿嘧啶衍生物的研究进展[J]. 化工学报,2010,28(11):653.
[5]Zielenkiewicz, W. Szterner, P. Kamin ski, M. Vapor Pressures, Molar Enthalpies of Sublimation, and Molar Enthalpies of Solution in Water of Selected Amino Derivatives of Uracil and 5-Nitrouracil. J. Chem. Eng. Data 2003, 48, 1132-1136.
[6]Tian, S. T. Zhang, Z. J. Zhang, Z. J. Chen, X. J. Xu, K. Z. How racil tautomers there are? Density functional studies of stability ordering of tautomers. Chem. Phys. 1999, 242, 217-225.
[7]Zhao, Y. Truhlar, D. G. J. Phys. Chem. A 2004, 108, 6908.
[8]Dunning, T. H. J. Chem. Phys. 1989, 90, 1007.
[9]M. J. Frisch, G. W. T., H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, A. G. Liu, Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez, and J. A. Pople. ; Revision B.05 ed.; Pittsburgh PA: Gaussian, Inc.: 2003.
[10]卢天,陈飞武. Bond Order Analysis Based on the Laplacian of Electron Density in Fuzzy Overlap Space[A].
[11]中国化学会.中国化学会第29届学术年会摘要集——第15分会:理论化学方法和应用[C]. 中国化学会,2014,1.
On the Influencing Factors of Dissolution Enthalpy of Uracil and its Derivatives
LÜ Hui-ping, ZHU Qi
(Department of Chemistry, Heze Shandong 274015, China)
Density functional at B3LYP/6-31+G* level was used to explore the enthalpies when uracil, 5- fluorine uracil, 5- chloro - uracil, 5- bromo - uracil and 5 - (triflu)- uracil is dissolved in methanol and water at 298.15 K and 1 atmospheric pressure. The calculated enthalpies of dissolution of uracil and its derivatives in water are in agreement with the experimental values, but they are very different in methanol. Therefore, the paper analyzes the molecular hydrogen bond, Laplacian of electron density and the influence of π of the molecules in two solvents. The results show that the hydrogen bond dissolved in methanol solution can be too large, the electron density value and Laplacian of electron density and π bond electron has good correlation with experimental results.
uracil; uracil derivatives; dissolution enthalpy; hydrogen bond; Laplacian of electron density; πbond
1673-2103(2017)02-0082-06
2016-12-02
吕惠萍(1985-),女,甘肃靖远人,助理实验师,硕士,研究方向:化学.
O641-3
A
猜你喜欢
中国民族民间医药·上半月(2023年1期)2023-06-27 04:05:06
国防科技大学学报(2020年6期)2020-12-07 09:25:48
核农学报(2020年2期)2020-03-11 08:33:16
测绘通报(2019年11期)2019-12-03 01:47:34
赤峰学院学报·自然科学版(2019年5期)2019-09-10 07:22:44
测绘学报(2018年1期)2018-02-27 02:23:07
现代计算机(2016年11期)2016-02-28 18:35:21
陕西师范大学学报(自然科学版)(2015年1期)2016-01-16 03:23:30
哈尔滨商业大学学报(自然科学版)(2014年5期)2014-09-14 04:32:28
中央民族大学学报(自然科学版)(2014年2期)2014-06-09 08:28:14
