
盐胁迫下大麦酵母双杂交文库的构建与鉴定
2020-01-02 06:49:02李颖波宗营杰刘成洪陆瑞菊黄剑华杜志钊许建华王亦菲
上海农业学报 2019年6期
李颖波,宗营杰,刘成洪,陆瑞菊,黄剑华,杜志钊,许建华,王亦菲*
(1上海市农业科学院生物技术研究所,上海市农业遗传育种重点实验室,上海201106;2上海市农业科技服务中心,上海200335;3光明种业有限公司,上海202171)
随着气候变迁和化肥的大量使用,我国农业生产中土壤盐渍化问题越来越突出[1]。盐胁迫显著抑制作物生长速度,降低分蘖和分枝数量,甚至致死,最终影响作物产量[2],已成为制约我国农作物生长与产量的重要因素之一。以往研究表明,植物的耐盐机制主要由渗透调节、离子平衡和抗氧化3种方式组成[2]。
受到高浓度盐离子胁迫后,一方面,植物在细胞质中积累多种次生代谢产物,如糖醇类、糖类、氨基酸及其衍生物、胺类和硫酸酯胆碱等,以增加细胞渗透能力,抵抗盐胁迫造成的细胞内渗透压改变[3];另一方面,植物通过细胞内的离子通道、质子泵和转运蛋白等相关蛋白的作用将Na+转运到液泡内隔离或外排,同时植物的抗氧化系统能清除盐胁迫下产生的大量活性氧、自由基等强氧化性物质[3]。目前研究发现,SOS基因家族、HKT基因家族和NHX基因家族在细胞Na+平衡上起着关键作用[4-7]。但是,植物对盐胁迫的抗性是一个复杂的生理过程,植物应对盐胁迫反应中许多信号途径仍有待阐明,研究盐胁迫下植物细胞中的信号途径和蛋白互作网络,将有助于提高对盐胁迫下信号转导途径的认识。相对于其他禾本科作物,大麦对盐的耐受性特别是组织耐受性较强,同时大麦也是遗传学研究中广泛应用的模式作物之一,且在国民经济生产发展中具有十分重要的地位。因此,研究大麦耐盐机制,不仅对我国大麦耐逆育种的进一步发展具有重要的理论意义和应用价值,也对研究其他禾本科作物的耐盐性具有重要的参考价值。
酵母双杂交是利用酵母遗传学分析蛋白质之间相互作用的方法,已广泛用于蛋白质组学、细胞信号转导和功能基因组学等领域[8]。酵母双杂交不但可以检测已知蛋白之间的相互作用,而且可以发现已知蛋白的互作蛋白,并且可以发现蛋白的新功能。近年来,研究者已在小麦、水稻、大麦、玉米等许多重要农作物中构建了不同组织、器官的酵母双杂交cDNA文库[9-11],为相关作物的蛋白质组学研究奠定了基础。本实验室前期获得了与盐胁迫相关的E3泛素连接酶基因,拟采用酵母双杂交技术筛选其互作靶蛋白,研究其在大麦耐盐反应中的作用通路。
本研究以盐胁迫下的不同大麦组织为材料,利用Gateway技术构建盐胁迫下的大麦酵母双杂交cDNA文库,以期为深入研究植物应对盐胁迫信号网络提供更多理论支持。
1 材料与方法
1.1 植物材料和盐胁迫处理
供试材料为‘花30’,是上海市农业科学院生物技术研究所细胞工程研究室育成的大麦品种。‘花30’种子浸泡12 h露白后,挑取发芽一致的种子播于盆钵中培养(昼/夜温度为22℃/20℃,相对湿度70%,昼/夜光周期为14 h/10 h)。当幼苗处于2叶1心期时,盆钵中加入400 mg/L的NaCl溶液进行处理。在不同的处理时间(0 h、24 h、48 h),分别剪取根、茎、叶,迅速置于液氮中冷冻,-80℃冰箱保存。
1.2 菌株和载体
大肠杆菌 DH10B、酵母菌株 Y187、文库载体 pDONR222、pGADT7-DEST均购自 Invitrogen公司(美国)。
1.3 主要试剂
Trizol Reagent、CloneMiner cDNA Library Construction Kit、FastTrack MAG mRNA Isolation Kit、UltraPureTMPhenol∶Chloroform∶Isoamyl alcohol(体积比25∶24∶1)购自 Thermo公司(美国);氨苄青霉素、卡那霉素购自Sigma公司(美国);DL2000 DNA Marker、Taq DNA Polymerase购自Takara公司(日本);琼脂糖凝胶DNA回收试剂盒、质粒小提试剂盒购自天根生化科技(北京)有限公司。
1.4 大麦总RNA的提取和mRNA的分离
参照李颖波等[12]方法提取盐胁迫后的大麦根、茎和叶的总RNA。将根、茎和叶的总RNA按照浓度1∶1∶2混合后分离mRNA。mRNA分离参照FastTrack MAGmRNA Isolation Kit说明书进行。分离的mRNA经质量及纯度检测后,用于后续的文库构建。
1.5 酵母双杂交初级cDNA文库的构建
初级cDNA文库构建参照 CloneMiner cDNA Library Construction Kit说明书进行。将纯化得到的mRNA进行cDNA第1链、第2链的合成及纯化,加重组接头attB1 Adapter。将cDNA利用Gateway技术与文库载体质粒pDONR222混合进行BP重组反应;重组反应产物经Proteinase K处理、纯化,利用电转化法转入大肠杆菌DH10B;加入1 mL SOC培养基,37℃培养1 h,即为cDNA初级文库菌液;加甘油至终浓度为20%(V/V),-80℃保存备用。
1.6 酵母双杂交次级cDNA文库的构建
次级cDNA文库构建参照CloneMiner cDNA Library Construction Kit说明书进行。将得到的cDNA初级文库菌液接种于含有相应抗性的LB液体培养基中,30℃过夜培养后抽提质粒,稀释质粒终质量浓度至300 ng/μL。取出1μL质粒,与文库载体质粒pGADT7-DEST混合进行LR重组反应;反应产物经Proteinase K处理、纯化,利用电转化法转入大肠杆菌DH10B。收集单克隆,加入3 mL SOC培养基,37℃培养1 h,即为酵母双杂交次级cDNA文库菌液;加甘油至终浓度为20%(V/V),-80℃保存备用。
1.7 酵母文库构建
将次级cDNA文库菌液接种于含有相应抗性的LB液体培养基中,30℃过夜培养后提取质粒。将5μg文库质粒用PEG/LiAc法转入酵母菌株Y187中,涂布于SD/-Leu平板上。于28℃培养3—6 d后收集单克隆,加甘油至终浓度为50%(V/V),-80℃保存。
1.8 文库库容量鉴定
分别从1.5、1.6和1.7转化后的文库原液中取10μL(酵母文库取100μL),稀释1 000倍(酵母文库菌液稀释10 000倍),吸出50μL(酵母文库涂100μL)涂布于含有相应抗性的LB平板上(初级文库使用卡那霉素抗性、次级文库使用氨苄青霉素抗性、酵母文库使用SD/-Leu平板),37℃培养12—16 h(酵母文库28℃培养2—3 d)后进行统计。文库滴度(CFU/mL)=克隆数×稀释倍数/涂板体积;文库总容量(CFU)=文库滴度×文库总体积。
1.9 文库插入片段长度和重组率鉴定
从各级文库LB抗性平板上随机挑取24个单克隆进行菌落PCR,初级文库菌落PCR使用的引物分别是 M13Forward(5’-TGTAAAACGACGGCCAGT-3’)和 M13Reverse(5’-TGTAAAACGACGGCCAGT-3’),次级文库和酵母文库菌落PCR使用的引物分别是pDEST22Forward(5’-TCGATGATGAAGATACCCCACC-3’)和pDEST22Reverse(5’-TCGATGATGAAGATACCCCACC-3’)。PCR反应条件:94℃预变性3 min;94℃ 30 s,56℃30 s,72℃3 min,30个循环;72℃延伸10 min,4℃保存。PCR产物用0.8%(W/V)的琼脂糖凝胶电泳检测,统计阳性克隆重组率,并鉴定插入片段长度。
2 结果与分析
2.1 大麦总RNA提取、m RNA纯化和dscDNA合成
提取NaCl溶液胁迫后的大麦品种‘花30’根、茎、叶的总RNA,用1.2%(W/V)琼脂糖凝胶电泳检测,可见28S和18S条带明亮清晰,表明该总RNA完整性好,未发生降解(图1A)。分离纯化混合后的总RNA样品得到mRNA,电泳检测mRNA质量,结果显示分离纯化的mRNA在较大范围内呈弥散状分布(图1B),表明mRNA质量良好。将mRNA反转录合成dscDNA,电泳显示dscDNA呈弥散状(图1C),片段分布较广。
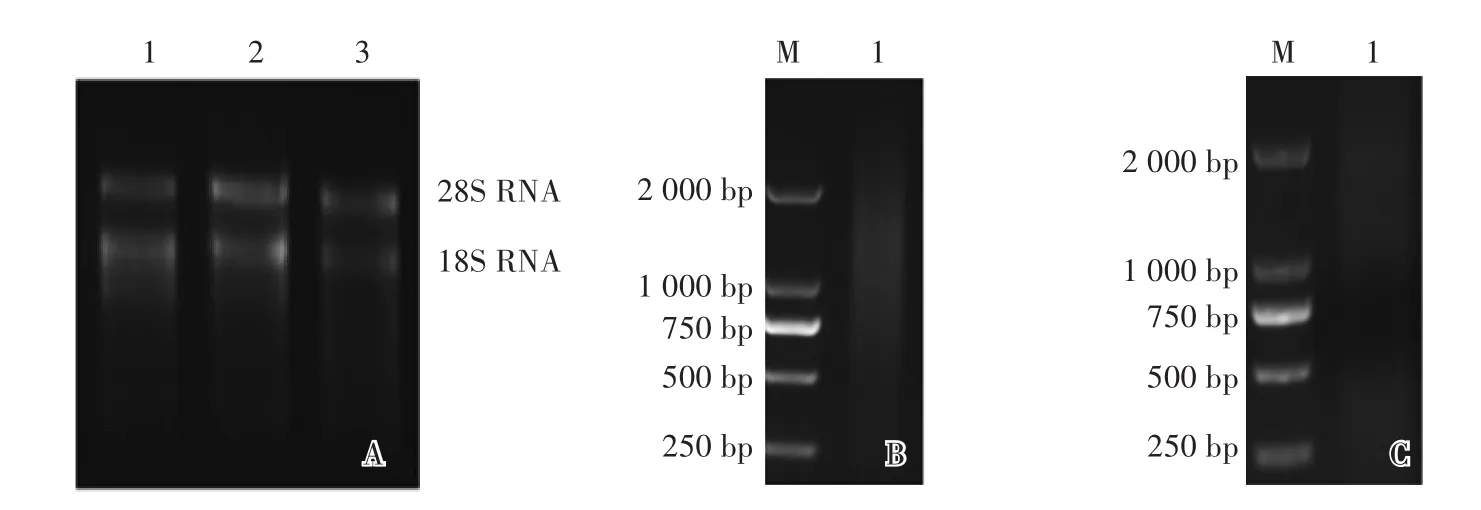
图1 大麦总RNA、m RNA和dscDNA电泳鉴定Fig.1 Electrophoresis of total RNA,mRNA and dscDNA of barley
2.2 初级cDNA文库构建和质量鉴定
将dscDNA和pDONR222载体重组,重组产物转入大肠杆菌中,获得初级cDNA文库。从初级cDNA文库中取10μL原始菌液稀释1 000倍,取50μL涂布于LB抗性平板上,37℃培养12—16 h后单克隆数为1 500个。经计算,文库滴度为3.0×106CFU/mL,初级cDNA文库总克隆数为1.2×107个。随机挑取24个单克隆进行菌落PCR,电泳结果显示,仅1个克隆呈阴性,其余为阳性克隆,重组率为95.8%,插入片段长度多为500—2 000 bp,插入片段平均长度大于1 000 bp(图2)。
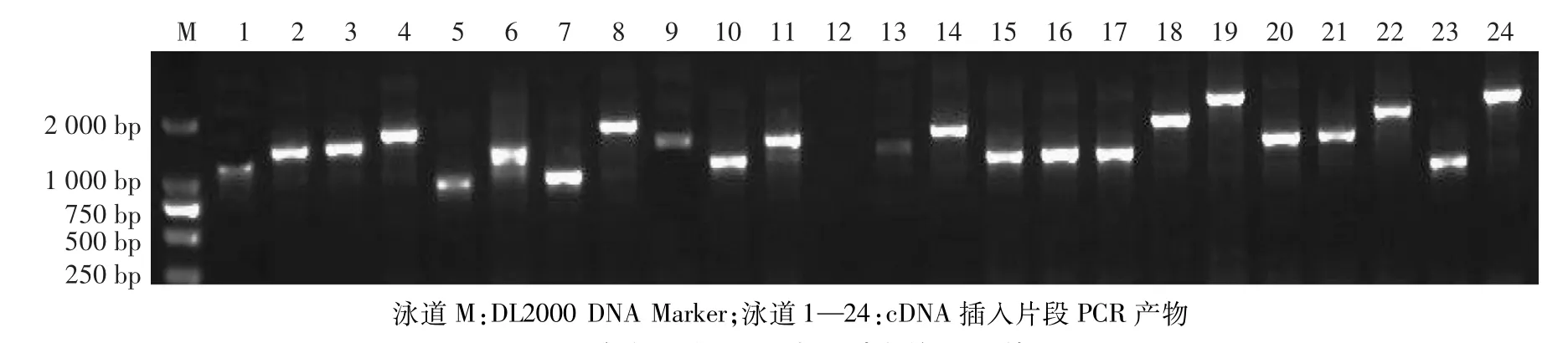
图2 初级文库cDNA插入片段的PCR检测Fig.2 PCR detection of inserted cDNA fragments in primary library
2.3 次级cDNA文库构建和质量鉴定
将初级cDNA文库摇菌提取质粒,并稀释至300 ng/μL。以pGADT7-DEST为目的载体,LR重组反应后转入大肠杆菌DH10B中,获得次级cDNA文库。从次级cDNA文库中取10μL原始菌液稀释1 000倍,取50μL涂布于LB抗性平板上,37℃培养过夜后的单克隆数为1 300个。经计算,文库滴度为2.6×106CFU/mL,次级cDNA文库总克隆数为1.04×107个。随机挑取24个单克隆进行菌落PCR,电泳结果显示,除1个克隆呈阴性外,其余均为阳性克隆,重组率为95.8%,插入片段多为500—2 000 bp,插入片段平均长度大于1 000 bp(图3),具有较好的多态性。

图3 次级文库cDNA插入片段的PCR检测Fig.3 PCR detection of inserted cDNA fragments in secondary library
2.4 出芽酵母双杂交文库的构建和质量鉴定
将次级cDNA文库摇菌提取质粒,取5μg转入酵母菌株Y187中,涂布于SD/-Leu平板上,收集单克隆获得酵母双杂交文库。从酵母文库原始菌液中取100μL,稀释10 000倍后涂布于SD/-Leu平板上,28℃培养2—3 d后单克隆数为700个。经计算,文库滴度为7.0×107CFU/mL。随机挑取24个单克隆进行菌落PCR,电泳结果显示全部为阳性(图4)。
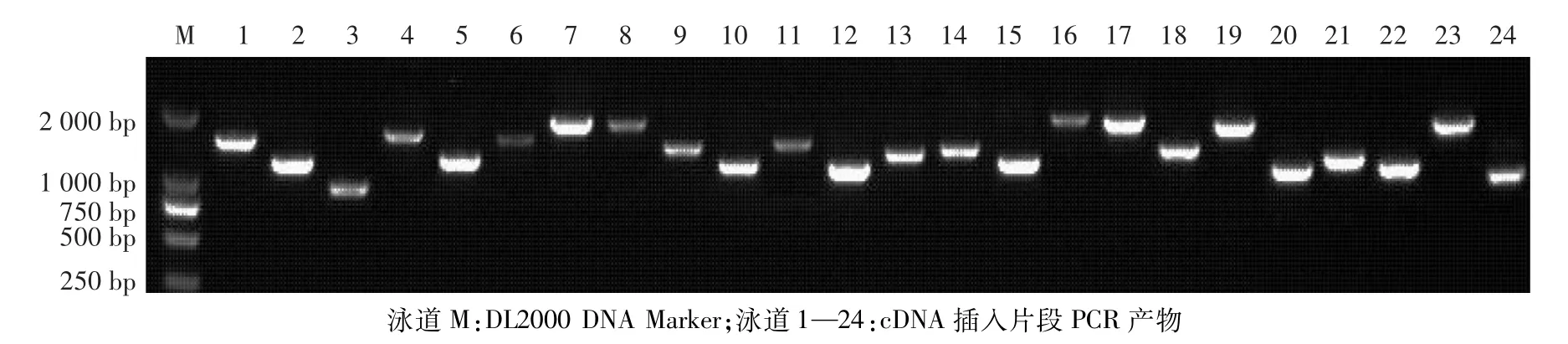
图4 酵母双杂交文库cDNA插入片段的PCR检测Fig.4 PCR detection of inserted cDNA fragments in yeast two hybrid library
3 讨论
高质量酵母双杂交文库是进行大规模互作蛋白筛选,获得阳性互作蛋白的前提[13]。本研究提取了盐胁迫后的大麦根、茎、叶的总RNA,并将其混合后用于构建酵母双杂交cDNA文库,防止了文库中组织特异性表达基因的丢失。
评价文库质量有两个重要指标,即cDNA文库的库容量和重组序列的完整性[14]。本研究分离纯化后得到的mRNA呈弥散状,具有较高的纯度和完整性,保证了后续文库构建质量。Clarke等[15]研究表明,满足低丰度mRNA筛选要求的最低文库滴定浓度为1.0×106CFU/mL。本研究中初级cDNA文库滴度为3.0×106CFU/mL,次级cDNA文库滴度为2.6×106CFU/mL,插入片段平均长度大于1 000 bp,表明所构建的cDNA文库覆盖度和和完整性良好,可满足常规文库筛选要求。酵母双杂交筛选主要有共转化和交配两种方法,其中交配法的效率相对较高[16]。本研究在次级cDNA文库的基础上,构建了大麦酵母双杂交文库,可以最大程度地保证筛选到所有的与靶蛋白具有相互作用的蛋白质分子。
猜你喜欢
作文小学高年级(2022年5期)2022-06-16 06:22:50
湖南农业大学学报(自然科学版)(2022年2期)2022-05-11 05:50:28
环境卫生工程(2021年4期)2021-10-13 06:52:26
作物学报(2021年11期)2021-08-31 05:37:08
猪业科学(2021年3期)2021-05-21 02:05:36
疯狂英语·新读写(2021年2期)2021-02-25 08:58:46
山东畜牧兽医(2021年6期)2021-01-11 10:31:57
幽默大师(2020年10期)2020-11-10 09:07:22
中华诗词(2019年1期)2019-11-14 23:33:56
猪业科学(2018年4期)2018-05-19 02:04:31
