
SDF-1/CXCR4及下游信号通路在骨性关节炎病程中的作用
2019-12-25 09:04何映红李彦林向耀宇陈泳佳杨骁
实用医学杂志 2019年22期
何映红 李彦林 向耀宇 陈泳佳 杨骁
昆明医科大学第一附属医院运动医学科(昆明655031)
近年来,骨性关节炎(osteoarthritis,OA)在老年人群体中的发病率逐年上升,且逐渐年轻化,传统治疗方法只能通过抑制炎症因子、改善关节腔微环境、置换关节等方式从症状及表观上间接治疗,但效果有限,未能从根本上阻断OA 发展。随着对骨性关节炎和类风湿性关节炎(rheumatoid arthritis,RA)的不断深入研究,发现基质细胞衍生因子-1/CXC 趋化因子受体4 型(stromal cell derived factor-1/ chemokine receptor-4,SDF-1/ CXCR4)信号通路在这两种疾病进程中发挥重要作用。OA 通常被称为退行性关节病,但这种说法并不完整,因为OA 不仅仅是一个磨损过程,而是由受累关节内的大量炎症介质驱动的关节组织异常重塑,致使关节软骨退化,同时软骨细胞不能修复关节软骨基质,导致组成和结构的变化。然而,关节软骨不是该疾病中唯一受影响的关节内组织。OA 软骨下骨通过间充质干细胞的增殖和软骨细胞的分化表现出重塑和微结构变化,使软骨下骨增厚、关节边缘中骨赘形成[1]。此外,免疫细胞渗入OA 滑膜,增强促炎介质的产生,这种滑膜炎也会导致软骨细胞基质失调,从而使软骨退化,半月板损伤及韧带退化[2],导致疼痛、畸形和功能丧失。而所有的这些过程都涉及有关信号分子的传递。目前的研究[3-4]表明,SDF-1/CXCR4 在骨性关节炎的进展中发挥重要作用,SDF-1 与其受体结合后使其C 末端的酪氨酸残基被磷酸化,激活多种下游信号通路,从而促进OA 的发生与发展。
1 MAPK 在OA 中的作用
磷酸特异性免疫印迹证实SDF-1α 激活软骨祖细胞中的所有三种丝裂原活化蛋白激酶通路(mitogen activated protein kinase pathway,MAPK),包括ERK1/2 通路,JNK 通路和P38 通路[5]。创伤后关节炎的研究发现SDF-1 可激活p38 和ERK1/2通路,它们是激活蛋白-1(activator protein-1,AP-1)活性的有效诱导剂,AP-1 对酒石酸抗性酸性磷酸酶(tartrate-resistant acid phosphatas,TRAP),基质金属蛋白酶(matrixmetalloproteinase-9,MMPs)和组织蛋白酶K(cathepsin K,CK)的表达至关重要[6],而TRAP 和CK 是破骨细胞的特异性标志物,也就是说,SDF-1 可以通过激活AP-1 增加破骨细胞的分化,促进骨质吸收,降低骨质钙含量,从而增加骨质疏松的风险[3]。在亚细胞水平,SDF-1 激活p38和Erk1/2 信号通路,其一方面诱导细胞周期蛋白D1 促进软骨细胞增殖,另一方面激活Runx2,增加MMP-13 生成的量,促进软骨细胞向末端成熟和凋亡[7]。颞下颌关节OA的研究证明激活ERK 信号通路有助于诱导基质金属蛋白酶的产生[8]。Erk1/2信号通路的激活可以诱导许多蛋白激酶级联反应,并将细胞外信号传递到细胞中[9]。磷酸化的ERK1/2 进入细胞核并触发转录因子和MMPs(包括MMP-3 和MMP-13)的活性,这有助于OA 病程启动和进展过程中细胞外基质的降解,引起一系列可调节细胞凋亡的细胞反应[10]。此外,p38 的激活,刺激软骨细胞分泌基质金属蛋白酶13(MMP-13),MMP-13 可降解人OA 中的Ⅱ型胶原蛋白和聚集蛋白聚糖,从而诱导软骨细胞死亡[11]。SDF-1 激活p38 通路使得血管内皮生长因子(vascular endothelial growth factor,VEGF)表达上调,促进滑膜血管形成,增加滑膜炎症因子和巨噬细胞浸润,加重OA 的炎症[5]。同时软骨及软骨下骨血管形成增加,加剧软骨细胞肥大及软骨下骨赘形成增加,加重OA 晚期关节退变的速度[12]。但软骨祖细胞的条件培养试验表明VEGF 表达基本上仅被p38 特异性抑制剂阻断,而不能被ERK 和JNK 特异性抑制剂阻断,所以软骨祖细胞中的VEGF 表达反应依赖于p38 活化,但不依赖于ERK 或JNK 通路激活[5]。MAPK 级联信号通路(ERK1/2 和p38)信号通路激活后,可以调节环加氧酶2的表达[10],COX-2促进花生四烯酸转化生成前列腺素2,并在关节中诱导关节炎发生[13],故PGE2 被认为是关节炎病症中炎性疼痛的主要原因[14]。
2 PI3K/AKT 在OA 发展中的作用
CHEN 等[15]研究发现:SDF-1α 激活CXCR4 受体并引起磷脂酰肌醇3-激酶(phosphatidylinositol 3-kinase,PI3K)/Akt 途径的激活,PI3K/Akt 在人OA滑膜成纤维细胞的IL-6 表达中起重要作用,该途径增强c-Jun 与AP-1 位点的结合并导致IL-6 的反式激活,使IL-6表达的上调,IL-6是3种主要的炎性细胞因子之一,和IL-1β和TNF-α一起,与炎症引起的软骨损伤有关,IL-6 通过作用于软骨生理学的合成代谢和分解代谢机制而对软骨造成损害[16]。PI3K 抑制剂能够有效抑制SDF-1a 指导的IL-6 产生,但MAPK抑制剂却无此抑制作用,结果进一步证实了PI3K/Akt 的显性失活突变体,抑制了SDF-1a对IL-6 产生,而不是通过MAPK 途径抑制IL-6 表达[15]。此外,IL-6 还被证明是促进破骨细胞分化的趋化因子[17]。
PI3K 还可能调节软骨细胞的凋亡:在坏死性凋亡过程中通过PI3K 的自磷酸化使混合谱系激酶结构域样蛋白MLKL 磷酸化导致MLKL 从细胞质转运至质膜[18],促使质膜碎片化,促进凋亡。然而,也有研究[19]证明PI3K/Akt 信号传导被认为是NF-κB 信号传导途径的主要上游元件,也就是说PI3K/AKT 是通过调节NF-kB 信号通路来增强软骨细胞凋亡的。细胞凋亡与OA 组织中软骨破坏程度和基质分解代谢高度相关[20]。与MAPK 级联信号通路一样,PI3K/AKT 信号通路激活后,可以调节环加氧酶2 的表达[10],促进PEG2 关节炎关节中诱导,加重关节疼痛[13,21]。同时研究证明,PI3K/AKT 信号通路和MAPK 级联信号通路(ERK1/2 和p38)激活后,可使活性氧(reactive oxygen species,ROS)生成增加,而诸多证据表明人类各种疾病中的细胞损伤导致氧化应激增加ROS 产生,这些影响是通过与MMPs 的相互作用介导的[22]。在OA中活性氧参与调节涉及软骨退化的生化因子的产生,它们可能通过激活MMPs减少基质成分合成直接或间接地对所有基质组分造成损害。
3 NF-κB 在OA 发展中的作用
在体外软骨细胞培养试验中也发现:SDF-1/CXCR4 轴可能是软骨细胞中核转录因子-κB(Nuclear Factor-κB,NF-κB)信号传导的其中一个主要上游调节因子[7]。活化B 细胞的NF-κB 家族在广泛的生物学过程中具有重要作用,包括细胞存活,增殖,分化,凋亡,衰老,炎症和免疫反应[23]。NFκB 信号通路通过各种作用广泛参与OA 病理生理学,并在衰老和炎症期间在OA 软骨细胞中被激活。NF-κB 途径是致细胞外基质分解代谢作用的中枢调节剂,介导软骨细胞的炎症反应中的关键事件并导致细胞外基质损伤和软骨侵蚀[24]。
NF-κB 途径对诱导各种炎症相关因子至关重要:肽聚糖和LPS(TLR-2 和4 的配体),以依赖于NF-κB 的方式分别刺激软骨细胞增加MMP-1,3 和13 的表达,以及上调NO 产生[25]。而NO 可以激活MMPs,增加蛋白多糖降解,减少IL-1 受体拮抗剂的产生并介导软骨细胞凋亡[26]。此外,根据剂量和特定的氧化还原衍生物的研究,NO 还可以对OA 进展产生负面和正面影响,特定的NO 衍生物维持NF-κB 活化以响应促炎刺激和其他形式的NO 抑制其诱导[27]。除此之外,NF-κB 通路还可诱导IL-6,IL-1,TNFα,COX-2 等软骨细胞表达炎症相关基因[28],这些诱导的细胞因子进一步激活信号级联反应。COX-2 参与导致PGE2 合成的连续酶促反应,PGE2 通过与关节组织中存在的EP2 和EP4 PGE2 受体结合,在软骨退化和OA 进展中起重要作用,导致软骨退化增加和OA 软骨中的软骨细胞肥大[29]。PGE2 还参与IL-1β 诱导的MMP-2 表达和活化[30],并诱导关节软骨细胞凋亡。在这些促炎因子中,IL-1β 和TNF-α 主要参与炎症,通过引发一系列导致分解代谢增加和软骨细胞肥大的事件在软骨降解中发挥重要作用。
在体外软骨细胞培养研究中还发现:SDF-1/CXCR4 激活NF-κB 信号传导,NF-κB 被激活后可产生细胞周期蛋白D1 和MMP-13 。此外,该试验表明SDF-1 激活p38 和Erk1/2 和NF-kB 信号通路,这些信号通路对软骨细胞的增殖和成熟有不同的影响。其中,p38 MAP 激酶和NFkB 对SDF-1 介导的MMP-13 诱导至关重要,而MEK/Erk 途径仅部分参与。相反,MEK/Erk 和NF-κB 在SDF-1 诱导细胞周期蛋白D1 中起关键作用,但p38 MAP 激酶途径的作用有限[7]。
4 JAK/STAT 在OA 发展中的作用
ANTONIO 等[31]分析了JAK/STAT 途径的SDF-1a 激活,发 现SDF-1 与CXCR4 结合 后,JAK2 和JAK3 与CXCR4 结合并且可通过转磷酸作用以Gai 非依赖性方式激活,证明JAK2 和JAK3 都被激活并与CXCR4 相关,这种激活使得STAT 家族转录因子的几个成员的募集和酪氨酸磷酸化成为可能。同时他们的研究表明JAK/STAT 途径的激活是CC 和CXC 趋化因子的一般信号传导途径。JAK/STAT 信号传导的激活对于使OA 进展为滑膜关节衰竭的慢性炎症状态永久化是至关重要的。该领域目前的研究揭示了在这些条件下调节异常细胞存活,细胞凋亡和基质金属蛋白酶基因活性的分子机制[32]。
人和牛的软骨细胞模型实验中发现,JAK/STAT 在调节组织金属蛋白酶组织抑制因子(tissue inhibitor of metalloproteinase,TIMP)和MMPs 中起重要作用,TMIP 在关节组织中广泛表达,能够抑制MMP,具有抑制软骨在吸收的潜力[33],软骨细胞通过JAK/STAT 调节MMP-TIMP 平衡,当信号通路受损,MMP-TIMP 失衡,MMP 表达过多,软骨退化加速,关节炎便会进展加快[34]。见图1。
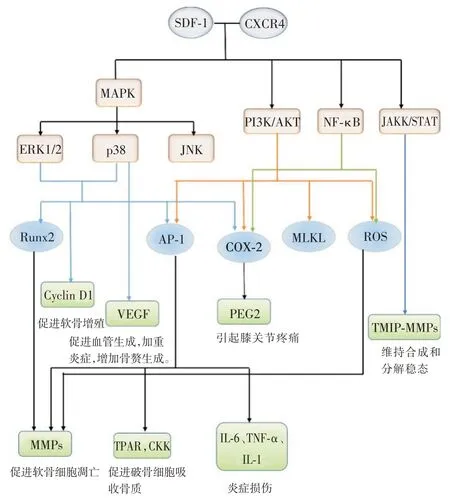
图1 SDF-1/CXCR4 及其下游信号信号通路传导图Fig.1 The graph of signal path in SDF-1/CXCR4 and the downstream signal
CXCR4 结合激活的下游信号通路并不是一一对应的关系,而是相互交错,互相影响。因此SDF-1与CXCR4 的结合后激活多种下游信号通路,涉及多种细胞因子,形成复杂的信号网络,可诱导运动、趋化反应、粘附,以及分泌基质金属蛋白酶、血管生成因子、白细胞介素、肿瘤坏死因子等,从而发挥生物学效应,调节细胞增殖,分化,存活和凋亡。SDF-1/CXCR4 信号通路与OA 中的各种病理事件有关。这些包括增加的炎症,血管生成和骨和软骨破坏,其参与风湿性疾病的发展。通过OA的体外和体内模型证明SDF-1 在这些相关过程中起关键作用。
5 总结与展望
鉴于SDF-1/CXCR4 信号通路在骨性关节炎的进展中具有重要作用,目前越来越多的学者将治疗骨性关节炎的目光放在了阻断SDF-1/CXCR4 信号通路上。因此,在已有几种疾病的临床试验中正在研究阻断CXCR4 受体的新治疗剂的功效和安全性,AMD3100 和TN14003 等阻断SDF-1/CXCR4通路的药物目前正用于治疗OA 的前期研究,这些药物作为治疗OA 的更为精准的靶向药物提供了一种很有前景的治疗方法。另外SDF-1/CXCR4 及其下游信号通路网络是一个庞大的信息库,与人体多种疾病相关,随着研究的不断深入,未来将会从组织、细胞、分子等层面系统深入研究SDF-1/CXCR4 信号通路调控骨性关节炎软骨退变效果及分子机理,OA 的病因和机制也会越来越明晰,这对于无论是OA 的早期诊断或是治疗都极大的进步,也为研发高效安全的骨性关节炎靶向治疗药物提供依据,从而使针对SDF-1/CXCR4 乃至其下游信号通路的拮抗剂将会有越来越多的发现,为临床治疗OA 提供全新的理念,丰富传统的单一的手术治疗OA 的方式。为延缓OA 进展提供新的思路,而OA 的治疗也将随着研究的进展更趋完善。
猜你喜欢
中国土壤与肥料(2021年5期)2021-12-12
疯狂英语·新读写(2021年10期)2021-12-07
中国土壤与肥料(2021年5期)2021-12-02
中老年保健(2021年10期)2021-08-24
北京园林(2020年4期)2020-01-18
中国临床医学影像杂志(2019年5期)2019-08-27
中国临床医学影像杂志(2019年1期)2019-04-25
中国医药指南(2017年3期)2017-11-13
中成药(2017年3期)2017-05-17
中华骨与关节外科杂志(2016年3期)2016-05-17
