
吡嗪酰胺-2, 5-二羟基苯甲酸共晶制备及过程在线研究
2019-12-21 09:03:38常芯瑗邓鸣峰王静康
天津大学学报(自然科学与工程技术版) 2019年1期
谢 闯,郝 菁,常芯瑗,刘 侃,邓鸣峰,王静康
吡嗪酰胺-2, 5-二羟基苯甲酸共晶制备及过程在线研究
谢 闯1, 2,郝 菁1,常芯瑗1,刘 侃1,邓鸣峰1,王静康1, 2
(1. 天津大学化工学院,天津 300072;2. 天津化学化工协同创新中心,天津 300072)
吡嗪酰胺(PZA)为最有效的抗结核药物之一,其水溶性差,通过形成共晶的方法可以改善其溶解度,提高药效.本文通过悬浮法制备了PZA和2,5-二羟基苯甲酸(2,5-DHBA)共晶,产品采用X-射线粉末衍射(PXRD)、差式扫描热分析(DSC)、傅里叶变换红外(FT-IR)和拉曼光谱(Raman)进行表征.结果表明,利用悬浮法可以制备出纯度较高的PZA-2,5-DHBA(1∶1)共晶.进一步采用衰减全反射傅里叶变换红外光谱仪(ATR-FTIR)和在线拉曼光谱等过程分析技术(PAT),监测并分析了该共晶的形成过程.结果表明,在30,℃下PZA-2,5-DHBA(1∶1)共晶在乙腈中的溶度积为9.4×10-4(mol/L)2;且该过程为PZA溶解-共晶生成的两步过程,存在成核介稳现象,过程控制步骤为共晶生长.
共晶;吡嗪酰胺;2,5-二羟基苯甲酸;悬浮结晶;过程分析技术
药物共晶是药物活性成分(API)与其他生理上可接受的酸、碱、盐和非离子化合物以氢键等非共价键形式结合在同一晶格中的新型化合物[1].共晶可以改变API的物理化学性质和药代动力学性质,具有重要的实际应用价值,因此受到了广泛关注[2].常用的共晶制备方法包括干磨法、湿磨法和溶液结晶法(包括冷却结晶法和悬浮法等)[3-5],其中溶液结晶法由于适合工业化大规模生产,是共晶制备的重要手段[6-7].常用的共晶表征方法包括X-射线粉末衍射法(PXRD)[8]、单晶衍射法(SCXRD)[9]、差式扫描量热法(DSC)[10]、热重分析法(TG)[11]、红外光谱法(FTIR)[12]、拉曼(Raman)光谱法[13]和固态核磁法(ssNMR)等[14].
过程分析技术(PAT)是通过实时原位测定原料、过程物料和过程本身的关键指标来设计、分析和控制整个生产加工过程,从而保证产品质量要求的一种技术手段[15].目前常用的PAT包括衰减全反射傅里叶变换红外光谱仪(ATR-FTIR)、拉曼(Raman)光谱仪、聚焦光束全反射测量仪(FBRM)和粒子成像测量仪(PVM)等.PAT为结晶过程的研究提供了方便快捷的技术手段,许多研究者利用PAT技术获得了重要的研究成果:杨金锁[16]利用FBRM等在线监测技术优化了巴比妥酸-尿素共晶的结晶工艺;Tong等[17]利用在线红外研究了乙水杨胺-糖精共晶的多晶型转化机理;Kulla等[18]利用在线拉曼研究了研磨法制备吡嗪酰胺-草酸共晶的形成过程.
吡嗪酰胺是重要的抗结核药物,属于生物药剂学分类系统(BCS)中的Ⅱ类药物,水溶性较低,通过形成共晶可以较好地改善其水溶性;2,5-二羟基苯甲酸(2,5-DHBA)是制药过程中常用的抗氧化赋形剂,也是一种常用的解热镇痛药[19],两者可以形成1∶1的共晶[20],该共晶在30,℃下的水溶性为31.2,mg/mL,是吡嗪酰胺(19.6,mg/mL)的1.6倍.共晶形成过程的研究对共晶的理论研究及实际应用都有重要意义,但是目前关于吡嗪酰胺相关共晶形成过程的研究还少见报道,因此本文制备了PZA-2,5-DHBA共晶并监测了其形成过程.我们在筛选实验中发现在乙腈中可以制备出PZA-2,5-DHBA共晶,乙腈是合适的共结晶溶剂,且乙腈中不含有羟基和羧基,对API与配体的红外特征峰干扰少,可方便地采用在线红外来监测该共晶的形成过程,因此选取乙腈为过程研究的溶剂.该研究为共晶的工业化提供了重要的理论和技术支持.
1 材料与仪器
PZA-2,5-DHBA共晶的不对称单元结构如图1所示.
1.1 药品与试剂
吡嗪酰胺(纯度99%,,上海麦克林生化科技有限公司);2,5-二羟基苯甲酸(纯度99%,,天津希恩斯生物科技有限公司);乙腈(分析纯,天津市江天化工技术有限公司);实验用水为去离子水.实验中所有试剂均直接使用,未做进一步纯化.
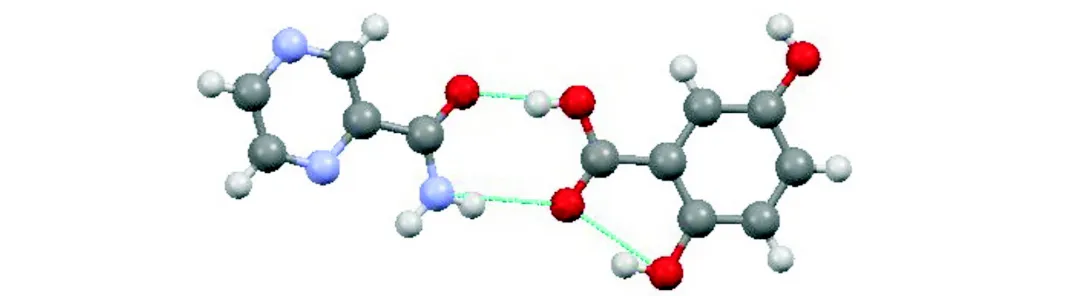
图1 PZA-2,5-DHBA共晶的不对称单元结构
1.2 仪 器
D/MAX 2500 X-射线衍射仪(Cu-Kα射线,=0.154,06,nm,日本岛津公司);DSC 1/500(美国梅特勒公司);ALPHA FT-IR红外光谱仪(德国布鲁克公司);RamanRXN2TM 分析仪(美国凯撒光学系统);ReactIR 15红外仪(美国梅特勒公司).
2 实验方法
2.1 共晶的制备
本实验采用悬浮法制备PZA-2,5-DHBA共晶:称取摩尔比为1∶1的PZA和2,5-DHBA于结晶器中,加入适量的乙腈,在30,℃下保持搅拌并反应3,h,然后过滤,烘干12,h得到PZA-2,5-DHBA共晶.
2.2 共晶形成过程研究
在线监测共晶形成过程时,采用分步加入反应物的方法.首先在一定量的乙腈中加入适量的2,5-DHBA,待2,5-DHBA完全溶解后,将在线红外和在线拉曼探头同时插入溶液中,再加入等摩尔量的PZA,整个过程中保持温度为30,℃.
3 结果与讨论
3.1 PZA-2,5-DHBA共晶的表征
3.1.1 PXRD光谱分析
图2给出了PZA、2,5-DHBA、悬浮产物实验测的PXRD图,以及利用PZA-2,5-DHBA单晶结构[20]模拟所得PXRD谱图.
由图2可知,悬浮产物的PXRD谱图中,PZA 在7.5°、15.9°、19.7°处的特征峰和2,5-DHBA在7.7°、15.2°、17.5°处的特征峰完全消失,但是在10°、14.5°、20.8°、27.16°、28.74°、29.2°处出现了新的特征峰,由此可知产物不是原料的物理混合(若为物理混合则衍射峰为两原料衍射峰的简单叠加),而是确实形成了新相(当产物和原料谱图中衍射峰位置不同时,说明产物中有新相形成):该实验所能形成的新相可能为两原料的多晶型,两原料的溶剂化物和PZA与2,5-DHBA形成的共晶.将该悬浮产物的谱图与PZA-2,5-DHBA单晶结构模拟计算产生的谱图做比较可知,两谱图特征峰位置基本相同,因此可以确定产物不是原料的多晶型或溶剂化物,可能为摩尔比1∶1的PZA-2,5-DHBA共晶.由于悬浮产物谱图中属于原料的特征峰完全消失,说明产物中原料含量较低,即生成了纯度较高的共晶.
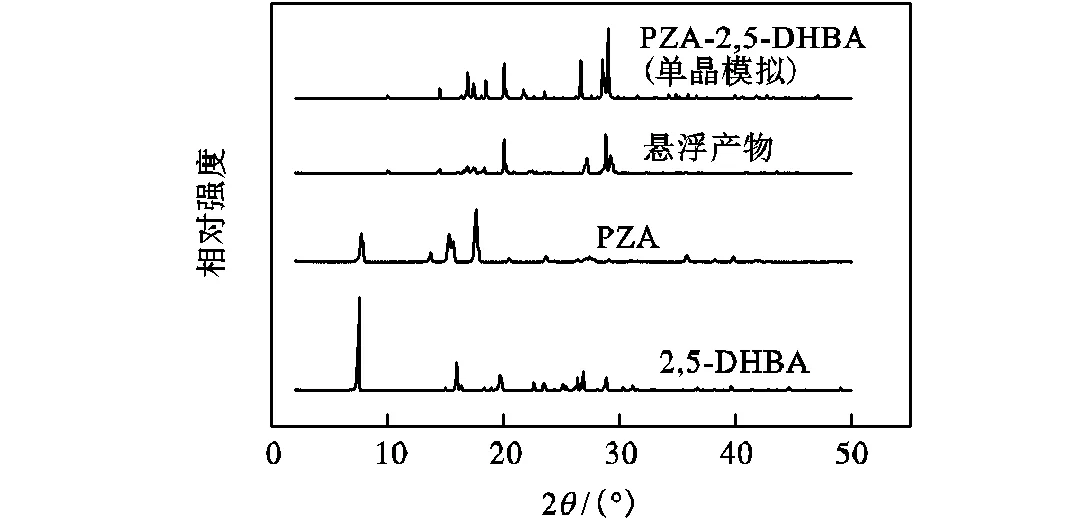
图2 PZA、2,5-DHBA以及悬浮产物的粉末衍射图
3.1.2 DSC热分析
熔点的测定也是判断是否生成共晶的重要手段之一[21],所测得的原料和产物的熔点如图3所示.
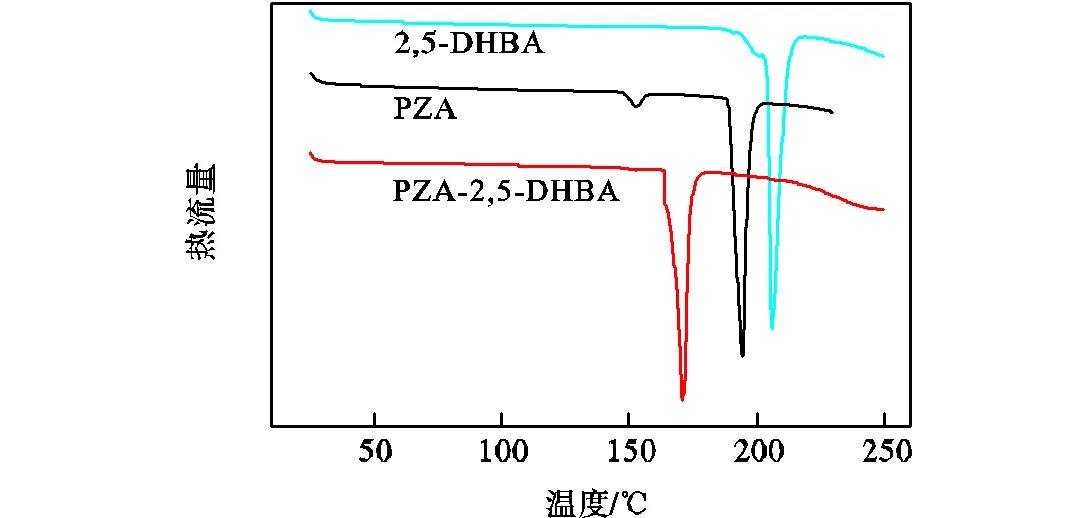
图3 PZA、2,5-DHBA和悬浮产物的DSC曲线
由图3可知,PZA在146,℃和190,℃处有两个吸收峰,其中146,℃的吸收峰为PZA的转晶峰,在此时PZA由α型转变γ型[22],而位于190,℃的吸收峰为γ型PZA的熔点峰;2,5-DHBA在202,℃有一吸收峰,该峰为2,5-DHBA的熔点峰;悬浮产物在164,℃处出现了新的吸收峰,该峰为悬浮产物的熔点峰.悬浮产物熔点低于两个纯组分的熔点,还可能为两纯组分的低共熔物,但由悬浮产物的PXRD谱图可排除(低共熔物谱图为两原料的简单叠加),证明悬浮产物确实为不同于两纯组分的新相:由于两原料的熔点峰在产物曲线中完全消失,且该产物熔点为164,℃,与文献[20]中由PZA-2,5-DHBA单晶测得的熔点一致,因此可判断该产物确实为PZA-2,5-DHBA共晶且纯度较高.
3.1.3 FT-IR光谱
如图4所示分别为PZA和悬浮产物在1400~4,000,cm-1范围内的红外谱图,其中1,702,cm-1处为PZA归属于酰胺基的—C=O基吸收峰,在悬浮产物中该吸收峰红移至1,669,cm-1处,这是因为PZA形成共晶后—C=O基与配体分子形成氢键使得羰基的振动频率降低从而导致特征峰向小波数方向移动;PZA中的—NH2分别在3,407,cm-1和3,285,cm-1处有两个吸收峰,悬浮产物中3,285,cm-1处的吸收峰红移至3,246,cm-1处,这是因为反应后—NH与PZA中的—C=O生成了N—H…O键导致振动频率降低,另一个—NH与PZA中的—OH生成了较弱的N—H…O键,对原—NH的伸缩振动影响较小,因此3,407,cm-1处的振动峰基本没有发生迁移.由此可知悬浮产物中确实有共晶中的氢键形成,从而进一步证明了PZA-2,5-DHBA共晶的形成.
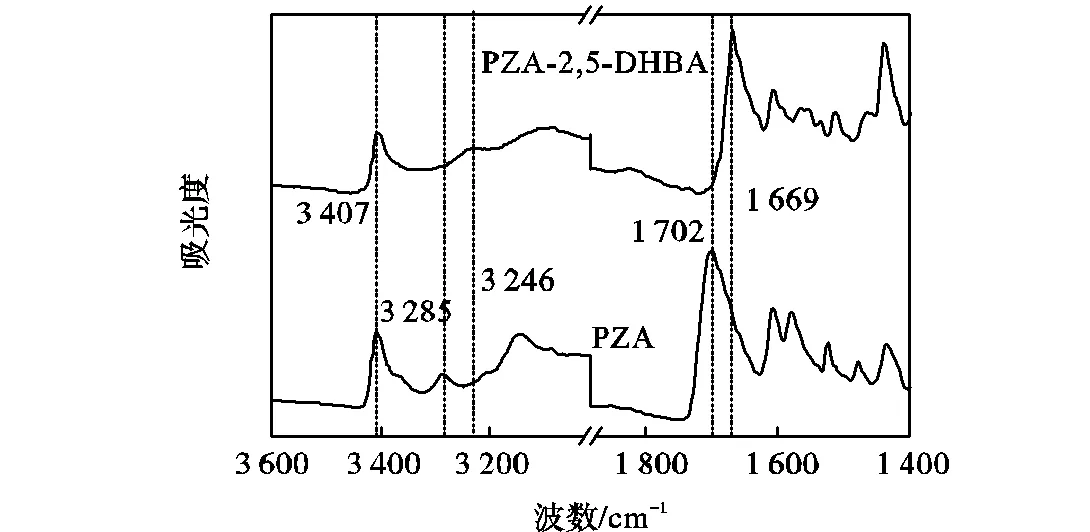
图4 PZA和PZA-2,5-DHBA共晶的FT-IR谱图
3.1.4 Raman光谱
图5给出了PZA、2,5-DHBA和共晶的拉曼光谱.由图可知,相对于共晶的拉曼谱图,PZA在501,cm-1、616,cm-1、1,384,cm-1、1,455,cm-1、1,487 cm-1、1,527,cm-1、1,576,cm-1、1,677,cm-1处的散射峰和2,5-DHBA在746,cm-1、1,200,cm-1、1,304,cm-1、1,352,cm-1处的散射峰消失,而PZA-2,5-DHBA共晶则在263,cm-1、1,186,cm-1、1,325,cm-1、1,564,cm-1、1,673,cm-1处出现了新的散射峰.其中,PZA在1,677,cm-1处的特征峰归属于—C=O基,在形成共晶后,由于形成氢键导致该散射峰迁移至1,673,cm-1处.由于PZA中归属于—NH2的特征峰位于3,429,cm-1处[23],该谱图无法体现共晶中—NH2的偏移情况.根据PZA-2,5-DHBA共晶拉曼峰的特征,可选取1,325,cm-1处这一强的新散射峰来监测其在制备过程中的变化.
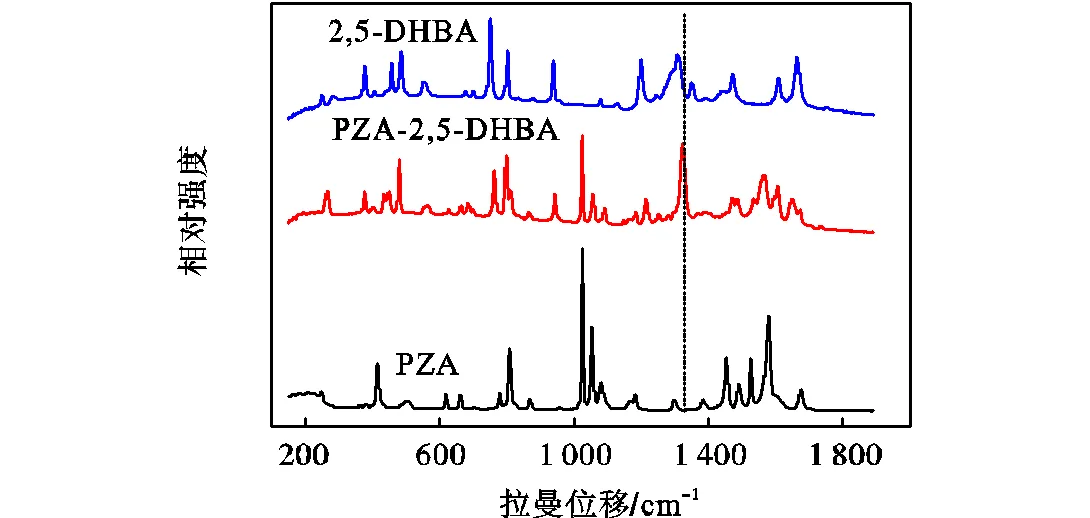
图5 PZA、2,5-DHBA和PZA-2,5-DHBA共晶的拉曼光谱
3.2 PZA-2,5-DHBA共晶形成过程研究
3.2.1 在线分析方法的确定
在线红外技术可以较为灵敏地监测共晶形成过程溶液中不同物质的浓度变化,实现对共晶形成过程的定性和定量分析,而在线拉曼可以与在线红外光谱相互补充,用于监测一些红外光谱无法检测到的信息,如溶液中固体颗粒的变化情况.因此,采用在线红外和在线拉曼相结合的方法可以实现对整个共晶形成过程的实时监控.
本文采用在线红外技术监测共晶形成过程中溶液的变化,实验进行前首先确定PZA和2,5-DHBA的特征峰.图6为乙腈、PZA和2,5-DHBA乙腈溶液的IR谱图:在乙腈中2,5-DHBA在1,190,cm-1处和1,692,cm-1处存在较强的特征峰,我们选取2,5-DHBA在1,190,cm-1处的特征峰来研究2,5-DHBA的浓度变化;PZA在1,702,cm-1处存在特征峰,由于两物质在1,692,cm-1和1,702,cm-1处的特征峰距离较近,当两物质混合后两峰会发生部分重叠,因此溶液中PZA的浓度变化需要通过间接计算1,702,cm-1处PZA的峰高得到.首先由2,5-DHBA在1,190,cm-1处(此处与PZA并无干扰)的特征峰标准曲线(标准曲线测定见下文)得到2,5-DHBA在不同时间下的浓度,然后由2,5-DHBA在1,692,cm-1处的标准曲线得到2,5-DHBA在该浓度处的峰高,则PZA所对应的峰高为两物质在重叠峰处的峰高减去2,5-DHBA在1,692,cm-1处的峰高,然后再根据PZA的标准曲线得到PZA的浓度;采用在线拉曼(1,325,cm-1处散射峰)检测悬浮液中固体颗粒的方法来监测反应形成的PZA-2,5-DHBA共晶的变化情况.
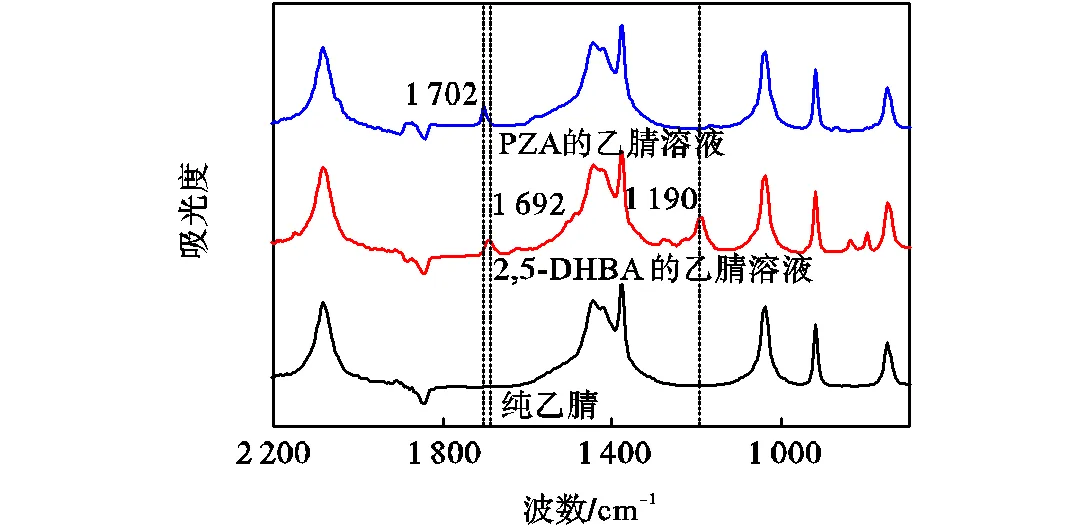
图6 PZA、2,5-DHBA乙腈溶液及纯乙腈的ATR-FTIR谱图
为了定量研究PZA-2,5-DHBA共晶形成过程,我们测定了不同浓度的2,5-DHBA乙腈溶液在1,190,cm-1和1,692,cm-1处的红外吸收峰高标准曲线,以及PZA在1,702,cm-1处红外吸收峰高对应溶液浓度的标准曲线(此处峰高为特征峰最大值处到特征峰两峰底的峰高即扣除基线之后的峰高),并对3曲线进行了线性拟合,拟合结果如图7所示,线性拟合参数如表1所示.由结果可知,在线红外光谱可以准确灵敏地检测到溶液中物质的浓度变化,利用这种方法研究共晶形成过程中溶液浓度的变化是可靠有效的.
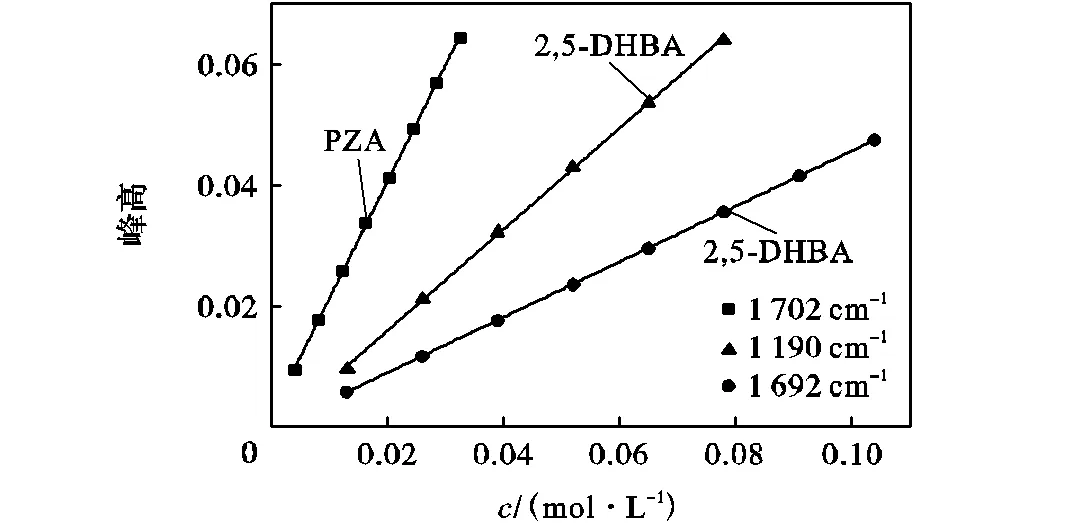
图7 PZA在1,702,cm-1、2,5-DHBA在1,190,cm-1和1,692,cm-1处ATR-FTIR峰高与浓度的标准曲线
表1 线性拟合参数
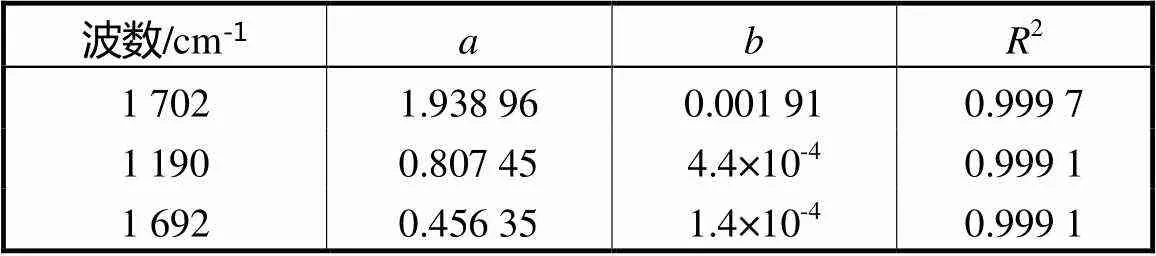
Tab.1 Parameters of linear fitting
注:、和2分别为斜率、截距和标准偏差.
3.2.2 在线监测共晶形成过程
根据在线红外信号及图7中的标准曲线,获得溶液中PZA与2,5-DHBA的浓度;根据在线拉曼信号反映所得共晶的情况;以上数据如图8(a)和(b)所示.该体系在本文实验条件的范围内仅发现了PZA-2,5-DHBA一种共晶,PZA与2,5-DHBA也未发现乙腈的溶剂化物,因此该三元体系的相图类型应如图8(c)所示.由图8(a)可知,2,5-DHBA的浓度在加入PZA的最初几分钟内迅速下降,然后下降速率变得缓慢,在约50,min后基本保持不变,维持在0.03,mol/L左右;同时,在线拉曼的监测信号也迅速增强,并基本与2,5-DHBA浓度同步趋于稳定,表明共晶的快速形成.在共晶的形成过程中,溶液中PZA的浓度总体逐步增大,并最终也维持在0.03,mol/L左右,与2,5-DHBA的浓度相同,这表明所加入的PZA晶体最终溶解完全,溶液与共晶达到固液平衡.溶度积sp可以体现固液平衡体系的溶解度大小,目前,溶度积概念已经被广泛用于共晶体系,Nehm等[24]测定了卡马西平-烟酰胺共晶在不同有机溶剂中的溶度积,Jayasankar等[25]测定了25,℃下不同比例的卡马西平-对氨基苯甲酸共晶在乙醇中的溶度积,Avdeef等[26]采用模型回归法预测了38种共晶的溶度积.在该体系共晶的固液平衡(式(1))中,其溶度积sp可由式(2)表示.根据实验平衡时的数据可知,在30,℃下该共晶在乙腈中的溶度积为9.4×10-4(mol/L)2.
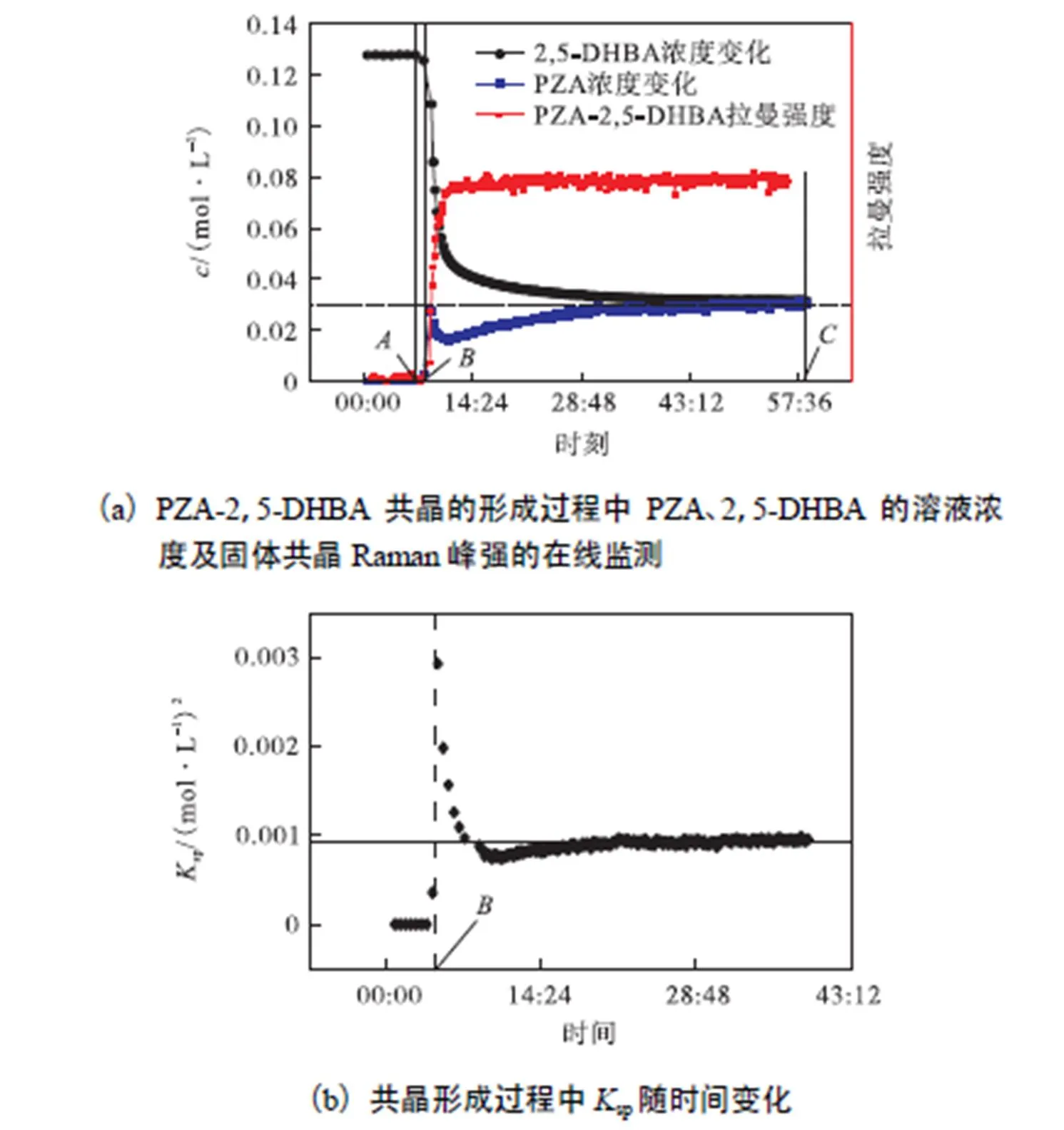

图8(b)给出了实验过程中sp的变化情况.由图可知,除开始阶段外,在共晶的形成过程中,溶度积几乎保持不变,表明成核后的共晶生成过程中,固液相基本接近平衡.值得注意的是,在加入PZA晶体后的开始阶段,sp一度超过平衡溶度积,达到0.003(mol/L)2,随后降回平衡溶度积.这表明出现了介稳现象,该现象从PZA浓度变化及共晶的在线拉曼数据也得到了印证:在该时间范围内,PZA浓度达到峰值后快速回落,同时,拉曼信号迅速增强,表明发生了成核过程.
综合以上结果,PZA-2,5-DHBA共晶悬浮实验的过程可分为以下3个阶段.①成核前:向2,5-DHBA的乙腈溶液中加入PZA晶体(点)后,随着PZA的溶解,系统点由点向点移动(见图8(c)),溶液中PZA的浓度及sp逐渐增大,2,5-DHBA由于在溶液中与PZA发生缔合但并未结晶,游离态的2,5-DHBA浓度降低;系统点到达点时,sp达到平衡溶度积,但由于介稳区的存在,共晶并未析出,PZA浓度和sp在成核前继续增大;②共晶成核:成核时,在线拉曼信号快速增强,PZA浓度和sp从峰值回落,同时2,5-DHBA浓度则加速降低;③生长阶段:随着共晶的增加,拉曼信号继续增强,sp则逐渐回归到平衡溶度积,由于消耗,2,5-DHBA浓度继续减小,而PZA浓度却经最低值后逐渐增大,但增速逐渐变慢.这是因为,成核导致溶液中PZA与2,5-DHBA的浓度降低,共晶的生成速度变慢,而晶体溶解则因PZA浓度的降低而加快;当PZA溶解速度超过消耗速度时,PZA浓度则通过最低点后继续上升;随着溶液中累积的PZA浓度逐渐增大,PZA晶体的溶解速度因此逐渐放缓,最终,随着PZA与2,5-DHBA的同步消耗,PZA晶体全部溶解,溶液中二者的浓度达到相等,并与共晶固体达到固液平衡的点(见图8(c)).该悬浮结晶过程是一个PZA晶体溶解-共晶生成的多步过程,从第3阶段PZA浓度逐渐上升的特征来看,该过程的速率控制步骤为共晶生长控制.
4 结 语
本文通过悬浮法制备了PZA-2,5-DHBA共晶,采用PXRD、DSC、FTIR和Raman光谱对悬浮产物进行了表征,其中PXRD结果表明悬浮产物与PZA-2,5-DHBA共晶为同结构晶体,DSC从热力学角度证明悬浮产物与PZA-2,5-DHBA熔点相同,FT-IR和Raman光谱从官能团角度证明悬浮产物形成了与PZA-2,5-DHBA一致的氢键,从而证明所得产物为PZA-2,5-DHBA共晶且纯度较高.利用在线红外和在线拉曼等PAT技术研究了该共晶悬浮制备的动态形成过程,结果表明:在30,℃下,PZA-2,5-DHBA共晶在乙腈中的溶度积为9.4×10-4(mol/L)2;该过程存在共晶成核介稳现象;该过程为PZA溶解-共晶生成的两步过程,其控制步骤为共晶生长.该研究可为药物共晶的在线监测及其定量分析提供理论和实验方法支持.
[1] Bolla G,Nangia A. Pharmaceutical cocrystals:Walking the talk[J]. Chemical Communications,2016,52(54):8342-8360.
[2] Duggirala N K,Perry M L,Almarsson Ö,et al. Pharmaceutical cocrystals:Along the path to improved medicines[J]. Chemical Communications,2015,52(4):640-655.
[3] Hasa D,Schneider Rauber G,Voinovich D,et al. Cocrystal formation through mechanochemistry:From neat and liquid-assisted grinding to polymer-assisted grinding[J]. Angewandte Chemie International Edition,2015,54(25):7371-7375.
[4] Bis J A,Vishweshwar P,Weyna D,et al. Hierarchy of supramolecular synthons:Persistent hydroxyl pyridine hydrogen bonds in cocrystals that contain a cyano acceptor[J]. Molecular Pharmaceutics,2007,4(3):401-416.
[5] 王灵宇,杜世超,董伟兵. 药物共晶多晶型的研究进展[J]. 化学工业与工程,2018,35(3):29-37. Wang Lingyu,Du Shichao,Dong Weibing. Research advances of polymorphism in pharmaceutical cocrystals[J]. Chemcial Industry and Engineering,2018,35(3):29-37(in Chinese).
[6] Lee M J,Chun N H,Wang I C,et al. Understanding the formation of indomethacin-saccharin cocrystals by anti-solvent crystallization[J]. Crystal Growth & Design,2013,13(5):2067-2074.
[7] Holaň J,Stěpánek F,Billot P,et al. The construction,prediction and measurement of co-crystal ternary phase diagrams as a tool for solvent selection[J]. European Journal of Pharmaceutical Sciences,2014,63:124-131.
[8] Solomon K A,Blacque O,Venkatnarayan R. Molecular salts of 2,6-dihydroxybenzoic acid(2,6-DHB)with N-heterocycles:Crystal structures,spectral properties and Hirshfeld surface analysis[J]. Journal of Molecular Structure,2016,1134:190-198.
[9] Janczak J. Supramolecular solid-state architectures formed by co-crystallization of melamine and 2-,3- and 4-chlorophenylacetic acids[J]. Journal of Molecular Structure,2016,1125:493-502.
[10] Hariprasad V M,Nechipadappu S K,Trivedi D R. Co-Crystals of ethenzamide:Study of structural and physico-chemical properties[J]. Crystal Growth & Design,2016,16(8):4473-4481.
[11] Sarma B,Saikia B. Hydrogen bond synthon competition in the stabilization of theophylline cocrystals[J]. Crystengcomm,2014,16(22):4753-4765.
[12] Castro R A E,Ribeiro J D B,Maria T M R,et al. Naproxen cocrystals with pyridinecarboxamide isomers [J]. Crystal Growth & Design,2011,11(12):5396-5404.
[13] Tong Y,Zhang P,Dang L,et al. Monitoring of cocrystallization of ethenzamide-saccharin:Insight into kinetic process by in situ raman spectroscopy[J]. Chemical Engineering Research & Design,2016,109:249-257.
[14] Wang N,Hao H,Lu H,et al. Molecular recognition and self-assembly mechanism of cocrystallization processes[J]. Crystengcomm,2017,19(27):3746-3752.
[15] 王 娜,陶晓龙,史欢欢,等. 过程分析技术在晶体多晶型研究中的应用[J]. 化学工业与工程,2017,34(2):1-9. Wang Na,Tao Xiaolong,Shi Huanhuan. Application of process analytical tools in polymorphism of crystalline materials[J]. Chemical Industry and Engineering,2017,34(2):1-9(in Chinese).
[16] 杨金锁. 巴比妥酸-尿素共晶多晶型及其共结晶过程研究[D]. 上海:华东理工大学化工学院,2017.Yang Jinsuo. Study on the Barbituric Acid-Urea Cocrystal Polymorphism and the Cocrystallization Process[D]. Shanghai:School of Chemical Engineering and Technology,East China University of Science and Technology(in Chinese).
[17] Tong Y,Wang Z,Yang E,et al. Insights into cocrystal polymorphic transformation mechanism of ethenzamide-saccharin:A combined experimental and simulative study[J]. Crystal Growth & Design,2016,16(9):5118-5126.
[18] Kulla H,Greiser S,Benemann S,et al. In situ investigation of a self-accelerated cocrystal formation by grinding pyrazinamide with oxalic acid[J]. Molecules,2016,21(7):917-926.
[19] 周石洋,陈 玲,杨善彬. 医药中间体2,5-二羟基苯甲酸的合成及表征[J]. 沈阳大学学报:自然科学版,2016,28(5):361-364.Zhou Shiyang,Chen Ling,Yang Shanbin. Synthesis and characterization of pharmaceutical intermediates 2,5-dihydroxybenzoic acid[J]. Journal of Shenyang University:Natural Science,2016,28(5):361-364(in Chinese).
[20] Mcmahon J A,Bis J A,Vishweshwar P,et al. Crystal engineering of the composition of pharmaceutical phases. 3. Primary amide supramolecular heterosynthons and their role in the design of pharmaceutical co-crystals[J].
Zeitschrift Für Kristallographie,2005,220(4):340-
[21] 毛会林. 药物共晶的制备与分析研究[D]. 天津:天津大学化工学院,2009. Mao Huilin. Study on the Preparation and Analysis of Pharmaceutical Cocrystals[D]. Tianjin:School of Chemical Engineering and Technology,Tianjin University,2009 (in Chinese).
[22] Castro R A E,Maria T M R,Évora A O L,et al. A new insight into pyrazinamide polymorphic forms and their thermodynamic relationships[J]. Crystal Growth & Design,2010,10(1):274-282.
[23] Sharma P,Nandi R,Gangopadhyay D,et al. Temperature dependent polymorphism of pyrazinamide:An in situ Raman and DFT study[J]. Spectrochimica Acta Part A Molecular & Biomolecular Spectroscopy,2017,190:177-180.
[24] Nehm S J,Rodríguez-Spong B,Rodríguez-Hornedo N. Phase solubility diagrams of cocrystals are explained by solubility product and solution complexation[J]. Crystal Growth & Design,2006,6(2):592-600.
[25] Jayasankar A,Reddy L S,Bethune S J,et al. Role of cocrystal and solution chemistry on the formation and stability of cocrystals with different stoichiometry[J]. Crystal Growth & Design,2009,9(2):889-897.
[26] Avdeef A. Cocrystal solubility product prediction using an in combo,model and simulations to improve design of experiments[J]. Pharmaceutical Research,2018,35(2):40.
Pyrazinamide-2, 5-Dihydroxybenzoic Acid Cocrystal:Preparation and in Situ Investigation
Xie Chuang1, 2,Hao Jing1,Chang Xinyuan1,Liu Kan1,Deng Mingfeng1,Wang Jingkang1, 2
(1.School of Chemical Engineering and Technology,Tianjin University,Tianjin 300072,China;2.Collaborative Innovation Center of Chemical Science and Engineering(Tianjin),Tianjin 300072,China)
Pyrazinamide(PZA),one of the most effective antituberculosis drugs,has poor aqueous solubility. By forming cocrystals,its solubility can be improved,which consequently improves its efficacy. In this study,PZA-2,5-dihydroxybenzoic acid(PZA-2,5-DHBA) cocrystal was prepared by slurry crystallization,and the product was characterized by powder X-ray diffraction(PXRD),differential scanning calorimetry(DSC),Fourier transform infrared spectroscopy(FT-IR),and Raman spectrometry.The results showed that the product of slurry crystallization was PZA-2,5-DHBA(1∶1)cocrystal of high purity. Furthermore,the slurry crystallization process was investigated using the process analytical technologies (PATs) of attenuated total reflectance-Fourier transform infrared (ATR-FTIR) spectroscopy and in situ Raman spectrometry. The solubility product of PZA-2,5-DHBA cocrystal in acetonitrile at 30℃ was found to be 9.4×10−4(mol/L)2. The slurry cocrystallization involved two steps:PZA dissolution and crystallization,and a metastable state occurred in nucleation. Moreover,the cocrystal growth was found to be the rate-controlling step.
cocrystal;pyrazinamide(PZA);2,5-dihydroxybenzoic acid;slurry crystallization;process analytical technology
TQ46
A
0493-2137(2019)01-0013-07
2018-03-15;
2018-04-07.
谢 闯(1980— ),男,博士,副教授.
谢 闯,acxie@tju.edu.cn.
国家自然科学基金资助项目(21776204).
the National Natural Science Foundation of China(No.21776204).
10.11784/tdxbz201803050
(责任编辑:田 军)
猜你喜欢
风流一代·经典文摘(2023年5期)2023-05-21 11:42:11
广东教育·高中(2020年10期)2020-11-16 06:07:30
数理化解题研究(2020年16期)2020-06-06 11:21:18
模具制造(2019年3期)2019-06-06 02:11:04
电子测试(2018年18期)2018-11-14 02:30:36
含能材料(2017年1期)2017-03-04 15:46:20
含能材料(2017年7期)2017-03-04 11:16:26
光学精密工程(2016年1期)2016-11-07 09:01:00
当代化工研究(2016年6期)2016-03-20 16:21:48
中学课程辅导·教学研究(2014年5期)2014-03-24 12:11:41
