
CRISPR/Cas 9介导的基质Gla蛋白基因敲除对破骨细胞分化的影响
2019-12-11 03:49赵俐婷王乃宁贺芳张燕
安徽医药 2019年12期
赵俐婷,王乃宁,贺芳,张燕
CRISPR/Cas 9(Clustered Regularly Interspaced Short Palindromic Repeats and CRISPR-Associated Proteins)系统是从原核生物中发现的一种防御外源性遗传物质入侵的自身免疫机制[1-2],能够识别自身和外源入侵DNA片段。它具有简单、方便、高效等特点[3-4],这种新型基因组靶向修饰技术已被广泛应用[5-8]。通过人工设计tracrRNA/cRNA复合体,形成单链向导RNA(guide RNA,gRNA),使Cas 9蛋白对特定的DNA序列进行靶向切割[4]。随后DNA通过非同源性末端接合或同源性重组,对断裂的DNA进行修复,发生替换、移码或缺失突变,达到基因敲除的目的[9-11]。
基质Gla蛋白(matrix Gla protein,MGP),是最初从牛骨骼中分离出的分子量为14 kD的维生素K依赖性循环蛋白[12]。MGP广泛存在于骨[13]、软骨[14]、牙质[15]等组织中,是调节骨形成和软组织钙化的重要基质蛋白[16]。MGP基因敲除小鼠出生后由于大动脉硬化破裂于6~8周内死亡[17-18]。MGP基因敲除小鼠还表现为软骨内骨化缺陷,尤其是生长板软骨,导致体型短小、骨量减少,提示MGP对成骨细胞分化有一定作用[19]。但迄今为止,尚未见到MGP对破骨细胞分化产生作用的报道。本研究于2018年5月至2019年1月利用CRISPR/Cas 9技术构建了MGP基因敲除的RAW264.7巨噬细胞系,为研究MGP在破骨细胞的功能提供依据。
1 材料与方法
1.1 材料
1.1.1 细胞 RAW264.7细胞系(购于中国科学院细胞库)用含10%胎牛血清(FBS)的α-MEM培养基进行培养,293-T细胞用含10%FBS的DMEM培养基进行培养,培养条件为37℃、5%二氧化碳,每隔2 d传代1次。
1.1.2 试剂 质粒提取试剂盒购于北京TIANGEN公司(批号Q5517)、DNA纯化试剂盒购于北京TIANGEN公司(批号R6308)、限制性内切酶BsmBI购于美国NEB公司(批号201805028)、T4连接酶购于西安海宁生物有限公司(批号AH70102A)、α-MEM培养基、DMEM培养基购于美国Hyclone公司、FBS购于美国GIBCO公司(批号42F9081K-2023-1)、巨噬细胞集落刺激因子(M-CSF)购于美国Pepro Tech公司(批号AF-315-03)、核因子κ B受体活化因子配体(RANKL)购于美国Pepro Tech公司(批号315-11C)、Lipofectamine2000转染试剂购于美国Thermo Fisher公司(批号11668027)。
1.2 方法
1.2.1 gRNA设计和寡核苷酸链合成 利用MIT在线软件设计针对小鼠MGP基因的gRNA,在靶序列正义链的5’端添加¬¬CACCG,反义链的5’端添加AAAC,3’端添加C。
1.2.2 gRNA表达载体构建 使用BsmBI限制性内切酶线性化质粒LentiCRISPRv2,酶切体系为:BsmBI 1 μL,LentiCRISPRv2 5 μg,10×buffer 5 μL,补水至50 μL,55℃酶切过夜。1%琼脂糖凝胶电泳对酶切质粒进行分离,利用DNA纯化试剂盒回收需要的片段。gRNA寡核苷酸单链退火形成双链,退火反应体系为:gRNA Up(100 μM)1 μL,gRNA Down(100 μM)1 μL,10×Buffer 1 μL,补水至 10 μL。95℃反应5 min后自然冷却至室温。将退火产物与线性化LentiCRISPRv2质粒连接,连接反应体系为:线性化LentiCRISPRv2质粒200 ng,稀释10倍的退火产物2 μL,10×Buffer 1 μL,T4连接酶1 μL,补水至10 μL,16℃反应4 h。将连接产物转化至感受态细胞,37℃过夜培养。挑取阳性克隆,摇菌,次日送公司测序鉴定。将含有正确序列的菌液保种,提取质粒备用。
1.2.3 包装重组慢病毒 293-T细胞接种于6孔板中,次日当达到70%密度时,利用Lipofectamine2000转染试剂,分别将800 ng LentiCRISPRv2-MGP-gRNA1,2,3,ctrl与600 ng psPAX2质粒、200 ng VSVG质粒一同转染入293-T细胞中。8 h后换新鲜培养基。再过24 h后收集病毒上清备用。
1.2.4 感染RAW264.7细胞及筛选稳定细胞株RAW264.7细胞接种于6孔板中,次日当达到50%密度时,加入含有50%病毒、8 μg/mL聚凝胺(polybrene)的新鲜培养基进行感染。24 h后换含有1 μg/μL嘌呤霉素的新鲜培养基进行筛选(感染成功的RAW264.7细胞具有嘌呤霉素抗性),3 d后收取细胞扩大培养。
1.2.5 实时荧光定量多聚核苷酸链式反应(qPCR)检测MGP基因敲除效率 提取细胞总RNA,取1 μg进行反转录,cDNA稀释10倍后使用TaKaRa实时荧光定量试剂盒,进行qPCR,反应体系为:2×mix 10μL、正/反向引物(10 μM)0.5 μL、cDNA 模板 5 μL,补水至20 μL。反应程序为:95℃预变性10 min,95℃变性10 s,60℃退火10 s,72℃延伸20 s,40个循环。使用甘油醛-3-磷酸脱氢酶(GAPDH)基因为内参,2-△△CT法进行数据分析。
1.2.6 蛋白质印迹法(Western Blot)检测MGP基因敲除效率 收集细胞,用RIPA细胞裂解液提取蛋白,BCA法进行蛋白定量。取20 μg蛋白进行凝胶电泳,转膜后用脱脂奶粉室温封闭1 h,一抗(1∶1 000)4℃孵育过夜,二抗(1∶1 000)孵育1 h,随后曝光分析灰度值。
1.2.7 qPCR检测破骨细胞分化标志分子的表达情况 对不同细胞克隆用破骨细胞诱导液(含有30 ng/mL M-CSF、100 ng/mL RANKL的10%FBS α-MEM完全培养基)进行诱导分化培养,每2天换液1次,5 d后待细胞出现明显的破骨细胞形态特征(多核、巨大),收集细胞,提取总RNA,具体操作见1.2.5所述,检测破骨分化标志分子Itgb3、Acp5、Ctsk的mRNA水平。
1.3 统计学方法 使用SPSS 17.0软件进行统计分析。数据以xˉ±s表示,多组间比较采用方差分析,组间两两比较采用Dunnett-t检验,以P<0.05为差异有统计学意义。
2 结果
2.1 LentiCRISPRv2-MGP-gRNA重组质粒的构建与鉴定根据小鼠MGP基因序列,利用网站评分选择3种gRNA,合成的序列如下。gRNA1:5′-CACCGTTGCCACGGCCAGCGCAGCC-3′,5′-AAACGGCTGCGCTGGCCGTGGCAAC-3′;gRNA2:5′-CACCGAAAGAGGTACTGGAACTCGA-3′,5′-AAACTCGAGTTCCAGTACCTCTTTC-3′;gRNA3:5′-CACCGGCTGTGTGAGCGCTACGCCA-3′,5′-AAACTGGCGTAGCGCTCACACAGCC-3′;对照序列:5′-CACCGCGCTTCCGCGGCCCGTTCAA-3′,5′-AAACTTGAACGGGCCGCGGAAGCGC-3′。将上述寡核苷酸退火后与LentiCRISPRv2质粒连接,构建成LentiCRISPRv2-MGP-gRNA重组质粒,测序鉴定结果如图1所示,表明重组质粒构建成功。
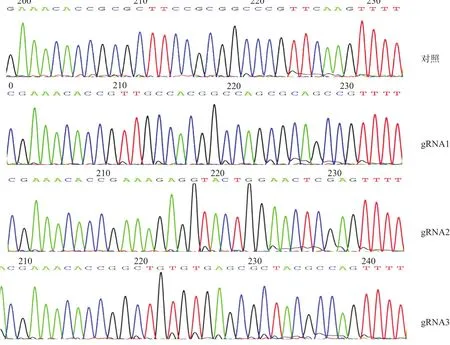
图1 LentiCRISPRv2-MGP-gRNA重组质粒测序鉴定结果
2.2 MGP基因敲除效果鉴定
2.2.1 MGP基因敲除效果qPCR鉴定 包装重组慢病毒并感染RAW264.7细胞,筛选后得到稳定MGP基因敲除细胞克隆,qPCR验证敲除效率。如图2所示,以ctrl组作为对照,LentiCRISPRv2-MGP-gRNA1细胞的MGP表达水平为对照细胞的(18±4)%,敲除了82%,其敲除效率最佳;LentiCRISPRv2-MGP-gRNA2细胞的MGP表达水平为对照细胞的(36±10)%,敲除效率为64%;LentiCRISPRv2-MGP-gRNA3细胞的MGP表达水平为对照细胞的(26±7)%,敲除效率为74%,差异有统计学意义(P<0.05)。
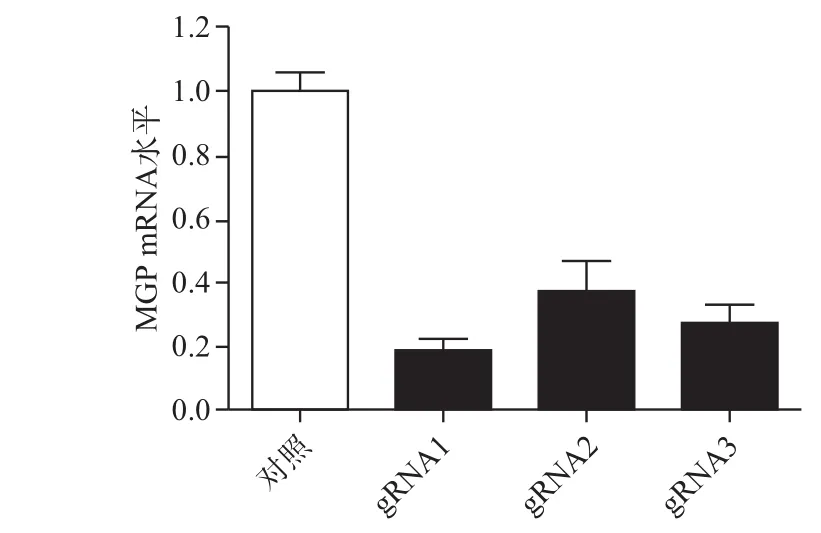
图2 不同RAW264.7细胞克隆MGP的mRNA水平
2.2.2 MGP基因敲除效果蛋白质印迹法鉴定 利用蛋白质印迹法检测MGP蛋白表达情况,验证不同gRNA敲除效果。如图3显示,以β-肌动蛋白(β-actin)为内参,与对照细胞(ctrl)相比,LentiCRISPRv2-MGP-gRNA1、2、3细胞克隆的MGP蛋白水平均明显降低,MGP基因表达被有效地人为干预。
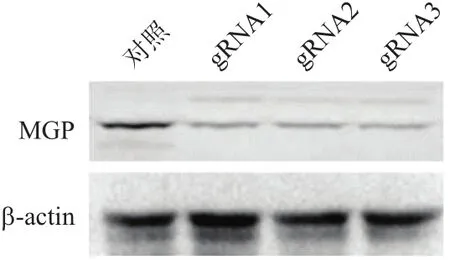
图3 不同RAW264.7细胞克隆MGP的蛋白水平
2.2.3 MGP基因敲除后对破骨细胞分化的影响对不同细胞克隆用含30 ng/mL M-CSF、100 ng/mL RANKL的10%FBS α-MEM培养基进行诱导分化,每2天换液1次,5 d后收集细胞,提取总RNA,反转录成cDNA,qPCR检测破骨细胞分化标志分子Itgb3、Acp5、Ctsk的mRNA水平。结果如图4所示,3种敲除MGP的不同细胞克隆经诱导分化后,与对照细胞(ctrl)[(1.10 ± 0.11)、(1.02 ±0.09)、(1.12± 0.04)]相比,LentiCRISPRv2-MGP-gRNA1细胞的破骨标志分子Itgb3、Acp5、Ctsk mRNA水平分别为(2.29±0.17)、(2.86±0.15)、(2.07±0.13),均显著增加(P<0.05);LentiCRISPRv2-MGP-gRNA2、LentiCRISPRv2-MGP-gRNA3细胞的破骨标志分子的mRNA水平也均显著增高[(1.79±0.12)、(1.75±0.17)、(1.69±0.09);(1.62±0.19)、(2.21±0.13)、(1.91±0.15),P<0.05]。以上结果提示敲除MGP后破骨细胞分化加速,表明MGP抑制破骨细胞分化。
3 讨论
CRISPR/Cas 9是一种在基因组水平上由RNA指导Cas 9蛋白选择性编辑目的基因的方法。由于操作简单、耗时少,应用日益广泛[1-4]。在基因敲除、调控基因的表达水平、疾病的基因编辑治疗等方面发挥重要作用。相较于其他基因编辑技术,CRISPR中Cas 9蛋白具有不需要形成二聚体来发挥作用,可通过构建多个gRNA实现多重编辑并同时沉默任意数量的单个基因等优势。
本实验利用CRISPR/Cas 9技术,在gRNA介导下使含有核酸酶、解旋酶等生物活性的Cas 9酶对MGP基因外显子特定的DNA序列进行特异性切割,通过非同源性末端接合或同源性重组,在细胞DNA修复作用下,达到敲除基因的目的[9-11]。本研究利用CRISPR/Cas 9技术将RAW264.7细胞中的MGP基因表达水平敲低82%,该稳定RAW264.7细胞系可用于进一步研究MGP功能。
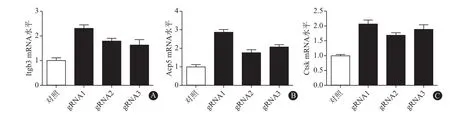
图4 不同RAW264.7细胞克隆破骨分化标志分子的mRNA水平:A为Itgb3,B为Acp5,C为Ctsk
MGP广泛存在于软骨、骨髓、动脉壁等组织中,是一种由84个氨基酸组成的维生素K依赖蛋白,调节骨形成和软组织钙化[20]。在骨骼中MGP参与了骨钙化调节[12]。有研究证实MGP是血管和软骨组织钙化的抑制剂,在血管钙化病变中MGP调节成骨细胞和软骨细胞的分化[17-18]。MGP可抑制核因子-κB(NF-κB)活性,可能通过抑制RANKL促进成骨细胞分泌骨保护素OPG,促进成骨细胞存活[19]。以上研究揭示了MGP在成骨细胞、软骨细胞中的作用,而MGP是否参与了破骨细胞分化成熟尚未见报道。
本实验通过CRISPR/Cas 9技术,建立了稳定低表达MGP的RAW264.7细胞系,利用蛋白质印迹法、qPCR对其MGP表达情况进行鉴定。相比于对照组,MGP基因的表达均明显降低。进一步对不同细胞克隆进行破骨诱导分化,并通过qPCR检测其破骨标志分子表达量,结果显示稳定低表达MGP的细胞克隆,其破骨标志分子的表达水平增高,证实MGP可抑制破骨细胞分化。但MGP参与破骨细胞分化的机制还有待深入研究,我们推测,MGP作为一种胞外基质蛋白,可能通过结合某些蛋白抑制破骨细胞分化。这需要更多的实验证据,将是我们近期工作的主要方向。
猜你喜欢
航天电子对抗(2022年4期)2022-10-24
环球时报(2022-09-20)2022-09-20
山东医药(2020年36期)2020-12-31
今日农业(2020年24期)2020-12-15
中国临床医学(2019年3期)2019-01-04
中国科技纵横(2018年2期)2018-11-29
中成药(2018年7期)2018-08-04
中国骨质疏松杂志(2016年1期)2016-01-29
小资CHIC!ELEGANCE(2015年15期)2015-09-01
现代检验医学杂志(2015年4期)2015-02-06
