
原位透射电镜技术在电池领域的研究进展
2019-12-11 04:59:08张利强唐永福刘秋男孙海明杨婷婷黄建宇
储能科学与技术 2019年6期
张利强 ,唐永福 ,刘秋男,孙海明,杨婷婷,黄建宇,
(1清 洁纳米能源中心,亚稳材料制备技术与科学国家重点实验室,燕山大学材料科学与工程学院,河北 秦皇岛066004;2燕山大学环境与化学工程学院,河北 秦皇岛 066004;3湘 潭大学材料科学与工程学院,湖南 湘潭 411105)
近些年来,锂离子电池(lithium-ion batteries,LIBs)发展迅速,并被认为是21世纪最理想的绿色能源之一[1-3]。自从1991年Sony公司制备出第一块商用LIB以来,LIBs经历了长足和快速的发展,已经在各种消费类移动电子产品中得到了广泛应用[4]。但是近期随着电动汽车等行业的快速发展,传统LIBs的能量密度已无法满足需求。为此,各类具有更高能量密度的电池应运而生,有Sn基和Si基LIBs、全固态金属锂电池、金属空气电池等[5-7]。然而,这些新型高能量密度电池在使用过程中存在着体积变化过大、易粉化等问题,导致电极材料破损和电池损坏[8-9],甚至造成严重的安全事故。因此,掌握电极材料在充放电过程中的结构演化过程对于理解电池的失效机理至关重要[10-11]。虽然已经过多年的研究,但LIBs电极材料在嵌锂、脱锂的过程中微观结构和相变机理还不是十分清楚,需要设计一种方法能在微观尺度上原位观察电极材料在充放电过程中的变化行为[12]。
一些重要的原位表征技术应运而生,主要包括:光学显微镜、扫描电子显微镜、原位X射线衍射、同步X射线吸收谱、同步拉曼光谱、傅里叶红外光谱、质量损失谱、核磁共振谱等[13-16]。其中,光学显微镜在电池循环时只能对微观结构演变信息提供低分辨率图像;扫描电子显微镜虽比光学显微镜有更高的分辨率,但仅能够反映材料的表面形貌和成分信息;原位X射线衍射可以提供极好的整体晶体结构信息,并且不需要在高真空环境,对于监测电化学反应过程十分有效,但空间分辨率较差,且不能提供微观形貌及成分变化信息。其他的无损分析方法,如傅里叶红外光谱、核磁共振谱、原子力显微镜、扫描隧道显微镜等亦被广泛应用,但各有优缺点。
透射电子显微镜(transmission electron microscope,TEM)早已被广泛用于材料科学中对结构的实时观察和颗粒的测量,但是应用原位透射电子显微镜技术(In situ TEM)研究电池仅是近十年才发展起来的[12,17-21]。这是因为传统的LIBs通常使用的是液体电解液,不能直接放入TEM高真空系统中。为此,HUANG等[22]成功地创造了第一个可在TEM高真空系统中运行的“开放型纳米电池”,设计概念完全不同于传统的密封液体窗方法。开放型纳米电池由单根纳米线作为负极、离子液体为电解液和LiCoO2正极所组成,装置利用了离子液体蒸气压极低的特点,可稳定存在于TEM的高真空环境中。开放型纳米电池的优势是:可以真正的实时和在原子尺度分辨率的条件下研究电池内部在充放电过程中的微观结构变化,除了直接观察形态演变的全过程,亦可通过电子衍射谱、电子能量损失谱等提供完整的结构和成分信息。
本文总结了应用In situ TEM技术在电池研究领域的重要进展,并且展望这项技术在电池方面的未来发展方向。
1 实验方法
在应用In situ TEM技术研究电池充放电行为的微观机理研究中,最大的困难在于如何在TEM中引入液态电解液。在传统研究中,大多使用原位液体反应池,其可以将液体与TEM的真空系统分离,采用这种方法的好处是可以选用任意类型的液态电解质,可以选择任何形状及形貌的电极材料,与真实电池的工作状态最为接近;但同时该方法由于上下表面都存在一层SiN隔膜,存在着操作困难、分辨率差、容易泄漏、难于成分表征等问题[12]。因此,设计开放型的电池测试系统用于In situ TEM表征至关重要。在2010年,HUANG等[22]首次将离子液体引入到透射电镜中,发现离子液体在电镜中可以稳定存在。在实验中,使用单根SnO2纳米线负极、离子液体电解液及LiCoO2正极构建成一个单纳米线LIBs[图1(a)],实时观察到了纳米线在充放电过程中的微观变化过程。
1.1 离子液体电解质纳米电池
实验中选择的液体电解质为离子液体电解液(ILE),是将LiTFSI溶解在P14TFSI中制备而成。在透射电镜中搭建纳米电池的方法是将一滴ILE滴在LiCoO2表面,而LiCoO2薄片是用导电银胶粘结在Al棒上;在另一侧,同样使用导电银胶将纳米线粘结在另一端Al棒上[图1(a)]。对于不同的电极材料,该实验装置都可确保良好的导电性能。实验中,由于离子液体的高流动性,最好选择具有较高长径比的纳米线。
在TEM中使用ILE的一个关键技术问题是防止电子束诱导ILE的退化。当电子束辐照剂量低时,ILE在实验中表现稳定。在高的电子束强度下,ILE快速失效并在纳米线表面形成胶状物包覆,导致Li+的运输能力下降,控制电子束辐射剂量一直是In situ TEM研究需关注的重要因素。此外,另一个困难是在电子束下ILE是不透明的,在纳米线浸入液体中的部分看不见,除非在实验中周期性的将纳米线从ILE中拔出。
1.2 固体电解质纳米电池
在使用固体电解质的实验中[图1(b)],纯金属Li是Li+的来源,在金属表面自然形成的Li2O作为固体电解质(仅允许Li+通过,电子难以通过)。由于实验中金属Li会短暂地暴露于空气中(约2 s),在新鲜的金属Li表面会形成一层Li2O。由于Li2O的带隙高达8 eV,是很好的电子绝缘体,为促进Li+穿过这层Li2O,在实验中需施加-2 V左右的过电势。虽然Li2O在传统的LIBs中很少被单独作为固态电解质,但其是固态电解质(solid electrolyte interface,SEI)层的重要组成部分。此外,前期研究成果显示Li+在Li2O中的扩散速率大约是10-10cm2/s,对于Li+的势垒仅有0.4 eV。众多实验已经证明Li2O固态电解质层的确是好的Li+导体,而非电子导体,并且应用此方法已在多种电极材料中成功进行纳米电池循环实验[23]。采用此固体电解质研究纳米材料的脱嵌Li行为有两个重要优势:首先,电极和电解质及其界面各部分在整个电化学过程中是看得见的;第二,适用于小尺寸纳米材料,比如小到几个纳米的纳米颗粒亦是可以进行In situ TEM充放电反应的,而这在离子液体纳米电池装置中是不可能实现的,因为ILE会将颗粒全部覆盖,无法成像。尽管目前应用此方法搭建的纳米电池与真实的电池相比还有很多不同,但此方法提供了材料在脱嵌Li过程中电极材料微观变化的最高分辨的结构和成分信息。同样需要注意的是,Li2O对电子束亦非常敏感,强电子束亦会把Li2O迅速分解为Li和O2。
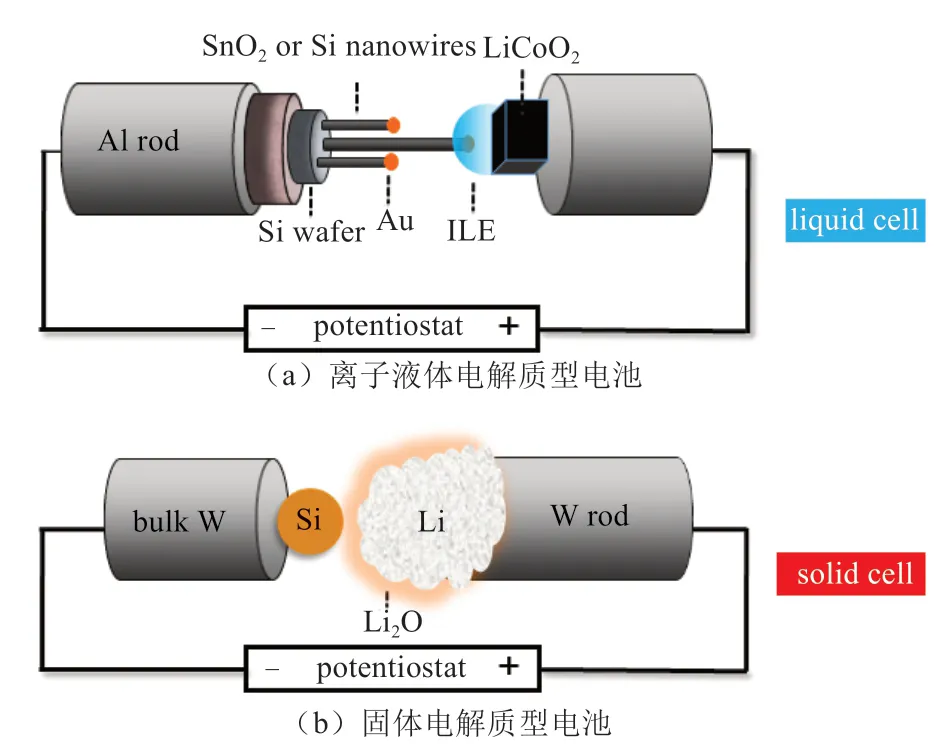
图1 应用In situ TEM 技术搭建的开放型纳米电池结构示意图[12]Fig.1 Schematic illustrations of the open cell nanobattery setup inside a TEM[12]
2 不同电极材料的In situ TEM 研究
2.1 Si 电极材料
Si基负极材料因具有很高的理论嵌锂容量(4200 mA∙h/g)而引起了科学家们的广泛关注[24]。但是,诸多研究发现在嵌Li过程中,Si基负极材料会产生显著的体积膨胀[25],并且在多次充放电之后,由于电极材料的粉化会导致电池容量快速衰减,因此限制了其在商用电池中的应用。虽然Si纳米颗粒已被广泛的用于制备锂离子电池电极材料,并且科学家们在缓解Si体积变化及保持循环稳定性方面进行了不懈的努力,但仍不清楚何种尺寸下Si纳米颗粒可以嵌Li而不产生裂纹和粉化。为深入理解Si纳米颗粒在嵌Li之后的体积变化行为,LIU等[26]应用In situ TEM技术对不同直径的Si纳米颗粒的嵌Li行为进行表征。
图2显示直径为940 nm的Si颗粒嵌Li时的体积和形貌变化行为。原始的Si颗粒是圆形的(图2),但嵌Li之后变为多边形。嵌Li行为是各向异性的,仅能在Si颗粒的某些晶面看到膨胀,Li+会优先沿着这些晶向嵌入。统计结果表明,Si颗粒的临界尺寸为150 nm(图3),低于这个尺寸的Si颗粒在嵌Li时没有裂纹产生,也不会断裂;若超过这个尺寸,Si颗粒首先在表面形成裂纹,然后由于嵌Li产生膨胀而断裂、粉化。表面断裂行为的出现归咎于嵌锂产生的拉伸应力(hoop stress)超过了Si材料的屈服强度,驱动了裂纹扩展。这些结果直接证明了小尺寸Si纳米颗粒在LIBs电池循环过程中的结构稳定性,这为宏观电池的设计提供了重要依据。
除了控制颗粒尺寸,掺杂和碳包覆(或者混合其他的导电材料)亦是宏观电池中最常见的电极材料改性的关键技术,被广泛的用于改善LIBs倍率性能和容量。原始Si由于导电性能差,其充放电速度非常低,为证实掺杂和碳包覆可有效提高电池的充放电性能,我们采用In situ TEM技术对比了不同类型的Si材料的充放电行为,主要包括:碳包覆Si纳米线,磷掺杂Si纳米线,碳包覆同时磷掺杂Si纳米线与原始本征(pristine)Si纳米线[27]。图4显示出碳包覆同时磷掺杂的Si纳米线在嵌Li后在反应前端产生很大的体积膨胀,形成核壳结构,并在首次嵌Li过程中显示超快的反应速度,纳米线变为螺旋形状,并且发现Si纳米线在嵌Li后变得非常柔软,与原始Si材料的易碎特性形成鲜明对比。
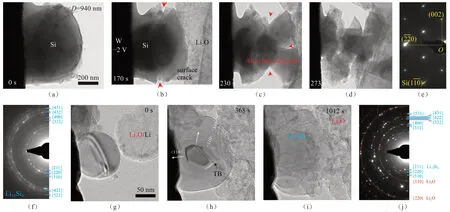
图2 (a)~(d)一个940 nm的Si纳米颗粒在嵌Li之后产生裂纹、破裂和粉化;(e)原始Si纳米粒颗粒为单晶;(f)嵌锂之后转为Li15Si4合金;(g)~(i)小尺寸Si纳米颗粒(约为150 nm)在嵌锂过程中产生均匀体积膨胀;(j)嵌锂之后同样由单晶转变为Li15Si4合金[26]Fig.2 (a) ~(d) A Si nanoparticle (NP) with a diameter of ~940 nm demonstrates obvious surface cracking during electrochemical lithiation.Electron diffraction patterns (EDPs) showing the phase transformation from single crystalline Si(e) to polycrystalline Li15Si4 after lithiation (f); (g) ~(i) When the diameters of Si NPs reduce to 150 nm, they demonstrate a uniform expansion without cracking and transform into Li15Si4 as well (j) [26]
通过碳包覆同时磷掺杂改善Si纳米线充放电性能结果总结如下(图5):①提高了充电倍率,碳包覆和磷掺杂都导致3~4倍电子导电率增加,最终导致充电倍率数量级增加(通过测算反应前端的移动速度),实验中观察到碳包覆同时磷掺杂Si纳米线具有最快的充电速度;②碳包覆同时磷掺杂的Si纳米线可以完全转化到Li15Si4相,具有最高的容量。这些原位实验结果再一次验证包覆和掺杂对于设计高能量和快速充放电的Si电极材料是大有前途的。

图3 Si 纳米颗粒的临界尺寸为150 nm,当粒径小于150 nm时,Si 纳米颗粒在第一次嵌锂过程中不会破裂,而当粒径大于150 nm 时,Si 纳米颗粒会发生破裂[26]Fig.3 Statistics showing the diameter critical size is around 150 nm.When the diameter size is below 150 nm, the Si NPs did not crack or fracture upon first lithiation.When D was larger than 150 nm, the Si NPs always cracked and fractured [26]
基于4种不同类型Si纳米线的电子衍射分析对比发现,Li15Si4是最终的嵌Li反应产物,这与许多宏观电化学测试结果一致,此外都观察到在嵌Li后Si材料的巨大体积膨胀。晶体Li15Si4相的形成是通过电子衍射所确定的,而非晶LixSi相的原子结构需要进一步深入研究。基于这些原位研究结果,计算得出每个Si原子最多可以存储3.75个Li,室温下Si电极材料的容量应该是3579 mA∙h/g,这与之前的理论预测是吻合的。LiCoO2构建一个单根纳米线LIBs(图6)。原始的SnO2纳米线是直的并且表面光滑,随着嵌Li反应的进行,反应前端沿着纳米线轴向由液体端向另一端运动,纳米线的直径和长度都发生了明显的变化,并由晶体相向非晶相转变。在 625 s时,纳米线快速发生扭曲,在纳米线表面形成珠子并且形成螺旋结构。研究发现,原始SnO2纳米线长度为16 µm,直径为188 nm,反应半小时后,纳米线长度变为约25.6 µm,直径约为272 nm,体积增大约240%。反应可以通过下面两个反应方程式表示:

图4 碳包覆和磷掺杂Si纳米线的超快充电反应。(a~i)施加偏压72 s内形貌的变化。由于反应速度超快,纳米线发生扭曲(反应前端标记红三角),反应前端平均移动速度为213 nm/s;(j)高倍TEM照片表明纳米线呈现核壳反应模式;(k~m)在嵌Li之后,同时碳包覆和磷掺杂Si纳米线的形貌变化。(k)反应之后,Si单晶纳米线变为晶体相Li15Si4(c-Li15Si4) 和非晶相a-LixSi共存;(l)嵌入Li后低倍的TEM照片;(m)高倍TEM照片发现纳米晶附着在非晶基体内,碳层在反应之后由3 nm增大到12 nm [27]Fig.4 Ultrafast charging of carbon-coated and phosphorusdoped silicon nanowires.(a~i) Morphology evolution in 72 s after the bias was applied.The spiral shape was observed only at very high lithiation rates (reaction front marked by red arrows).The average speed for the reaction front was 213 nm/s.(j) High magnification time-lapse TEM images showing an immediate expansion after lithiation and the core shell lithiation behavior.(k~m) Microstructure of the C-coated and P-doped Si nanowire shown in (j) after lithiation.(k) EDP showing coexistence of the crystalline Li15Si4 (c-Li15Si4) and a-LixSi phases.(l) Low magnification image of the lithiated part.(m) High-magnification image showing the contrast of nanocrystals dispersed in an amorphous matrix.The carbon layer was also swelled from originally 3 to 12 nm after lithiation [27]
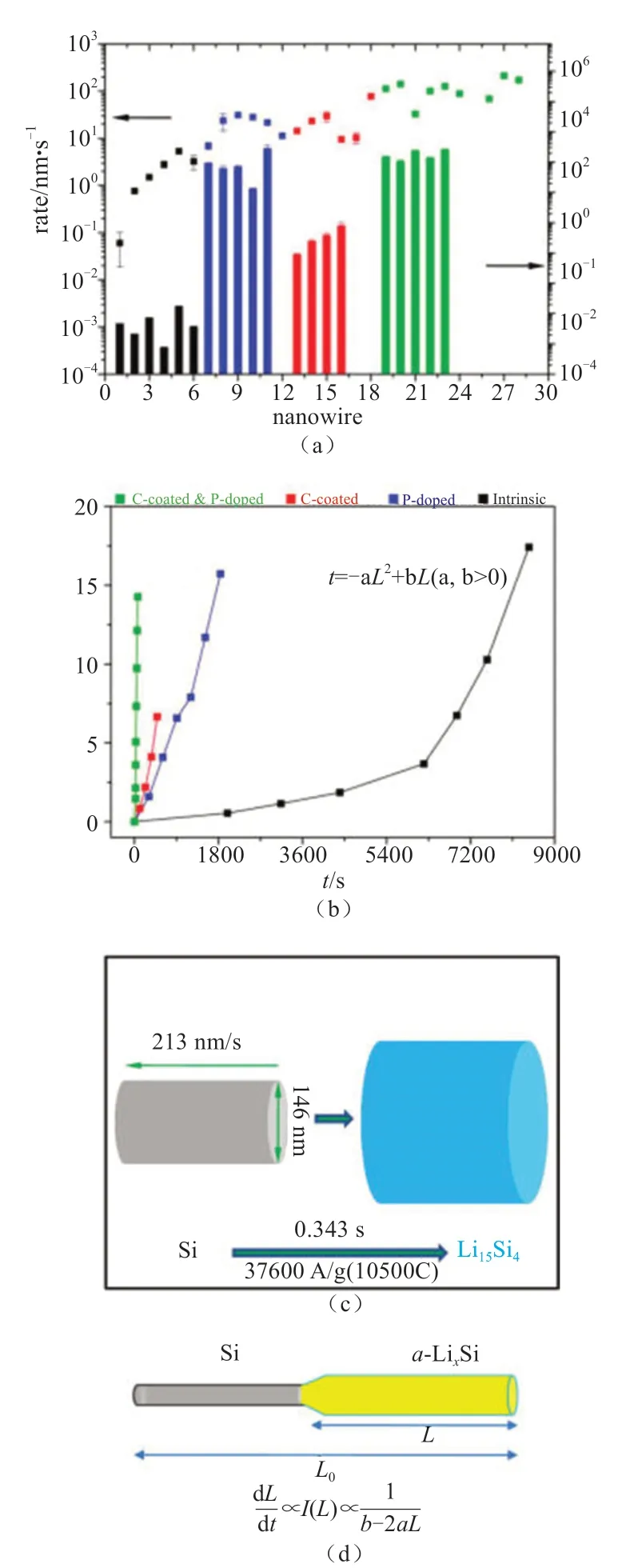
图5 (a)本征Si纳米线(黑色)、磷掺杂Si纳米线(蓝色)、碳包覆Si纳米线(红色)、磷掺杂同时碳包覆(绿色)Si纳米线的嵌Li速率和导电性比较;(b)不同类型Si纳米线反应距离与时间的关系图。对于本征Si纳米线,随着反应速度的加快,反应距离和时间成抛物线增加。对于其他三类改性的Si纳米线,反应距离和时间成线性关系,表明充电过程是由界面迁移控制,而不是由扩散控制;(c)磷掺杂同时碳包覆Si纳米线超快嵌Li反应过程示意图;(d)本征的Si纳米线缓慢嵌Li反应过程示意图[27]Fig.5 (a) The reaction front propagation speed and conductivities of the intrinsic, P-doped, C-coated, and C-coated on P-doped Si nanowires; (b) The reaction front migration distance vs time curves for these four types Si nanowires.The curve was parabolic for the intrinsic Si nanowire, with increasing lithiation speed as the reaction proceeded.The curves were linear for the other three types of Si nanowires,indicating the lithiation was interface reaction controlled rather than diffusion controlled; (c, d) Schematic illustration showing the lithiation for C-coated on P-doped Si nanowires and intrinsic Si nanowires[27]
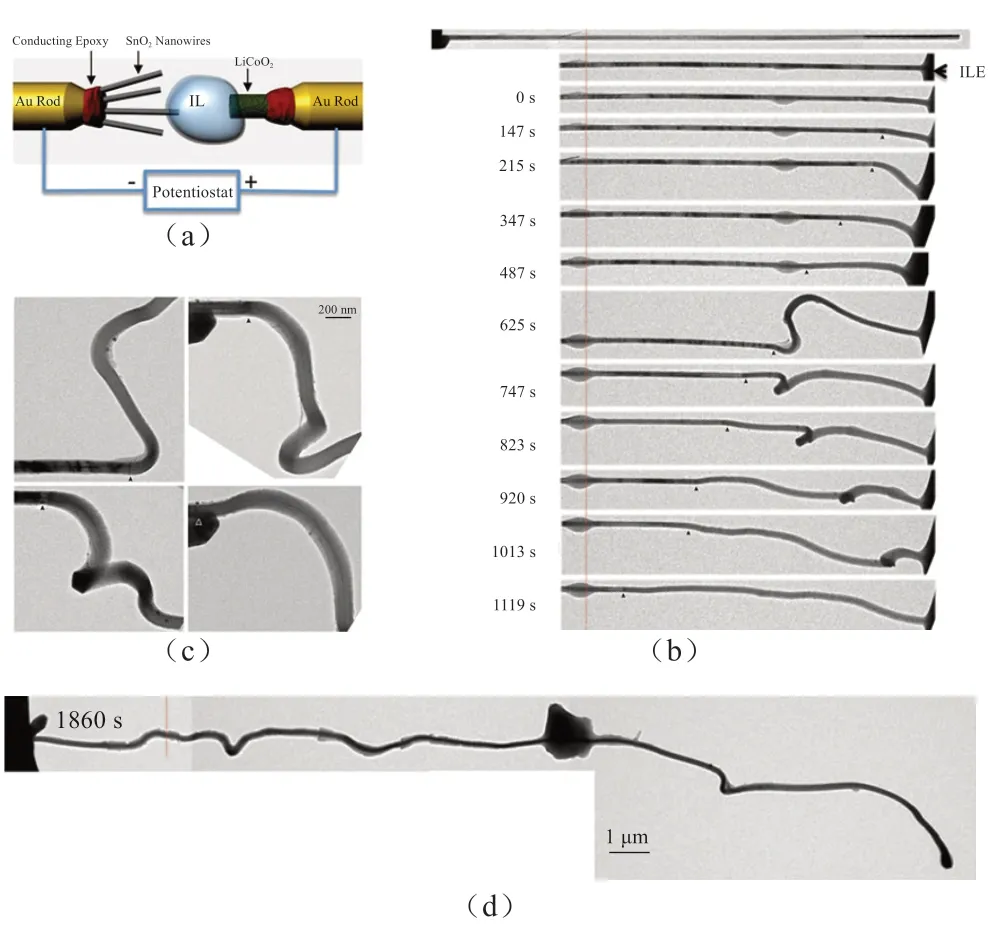
图6 (a)原位实验装置示意图;(b)原始的SnO2 纳米线是直的,随着嵌Li 反应的进行,纳米线发生了扭曲和显著的体积膨胀;(c)反应前端的高倍TEM 照片;(d)纳米线上黑色的颗粒为凝固的电解液[22]Fig.6 (a) Schematic of the experimental setup; (b) The initially straight nanowire became significantly twisted and expanding after lithiation; (c) are sequential high magnification images showing the progressive migration of the reaction front; (d) The big dark particle in the middle of nanowire is a polymerized IL[22]
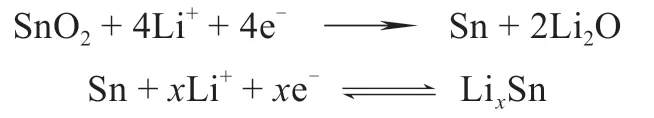
2.2 SnO2负极材料
除了Si材料,SnO2理论容量也高达781 mA∙h/g,是石墨的2倍,亦是未来LIBs领域最有希望替代碳材料的阳极材料之一[28-29]。HUANG等[22]在TEM中,使用单根SnO2纳米线、离子液体电解液及
电极材料在嵌Li过程中产生如此大的体积膨胀计算其内部会产生高达2 GPa的应力,如此大的应力往往会造成电极材料破裂或粉化[22],但由于此实验选择的是具有优异力学性能的纳米线材料[30],避免了此现象的发生。在宏观电池研究中,已发明了多种技术可缓解电极材料的体积膨胀,如采用中空或者多孔结构,或复合弹性缓冲层等[31-32]。嵌Li后造成的应变和体积膨胀是众多高能量密度电极材料共同面临的重要问题,通过包覆控制嵌Li所造成的应变可以改善许多高能量密度电极材料的循环稳定性。此外,在充放电过程中,控制电子和Li+在LIBs中的传输速率与提高电极材料的力学性能亦是制备高性能LIBs的重要途径[33-36]。因为SnO2材料的禁带宽度为Eg=3.6 eV,属于宽禁带半导体,导电性较差[37]。诸多宏观研究发现镀碳的SnO2纳米线材料与原始的SnO2纳米线相比,具有更高的容量和更长的循环寿命[29],但碳层提升LIBs效率的微观机理还不被人们所认知。
为此,ZHANG等[38]采用In situ TEM表征技术,证实了通过碳包覆可以改善SnO2纳米线的脱嵌Li行为(图7)。正如理论预测,研究发现碳包覆SnO2纳米线比未包覆的具有更快的嵌Li速度,与Si纳米线原位实验结果一样。在SnO2纳米线嵌Li+过程中,Li+有两条传导路径:一个是通过薄的表面ILE层,另一个是嵌Li后的纳米线。涂层的包覆导致Li+沿径向方向从ILE层表面快速扩散到纳米线内部;另一方面,涂层也有效抑制了纳米线沿径向的膨胀。包覆实验结果证明嵌Li所造成的体积膨胀是可以控制的,保护层的添加不仅可提高电池的倍率性能和容量,而且可改善嵌Li所造成的应变,这对于设计高能量密度电池电极材料具有重要的参考价值。
此外,原位实验发现最大的体积变化出现在首次嵌Li的过程中,同时表面包覆层的断裂亦发生在首次嵌Li过程中,体积变化在后来的循环中变得不明显,这意味着包覆层在首次嵌Li后即使发生断裂,仍然能显著地改善其循环性能,克服了首次嵌Li过程所造成的电化学冲击[38]。
3 金属空气电池的In situ TEM 研究
3.1 锂金属-空气电池
在近期各种新型电池的研究中,锂氧气电池由于具有超高的理论比能量(3458 W∙h/kg)而广受关注[39-40]。然而,缓慢的动力学反应过程、较高的过电位以及严重的副反应阻碍了锂氧气电池的商业化应用。为揭示锂氧气电池失效的根本原因,LUO等[41]使用环境透射电子显微镜(environmental transmission electron microscopy,ETEM)在氧气环境中,将金属锂与碳纳米管(carbon nanotube,CNT)组装成一纳米电池,原位研究Li-O2电池在充放电过程中微观结构的变化(图8)。研究发现CNT上发生的氧还原反应(ORR)最初产物为LiO2,随后转变为Li2O2和O2。O2的释放导致了在三相接触点纳米球的形成,其中Li2O为外壳,内部为Li2O2。研究还发现,释放O2方式与锂离子扩散和电子传输路径是密切相关的。通过In situ TEM表征,掌握了Li-O2电池在充放电过程中的微观结构变化,这对于理解电池的工作原理和探索其稳定性具有重要意义。
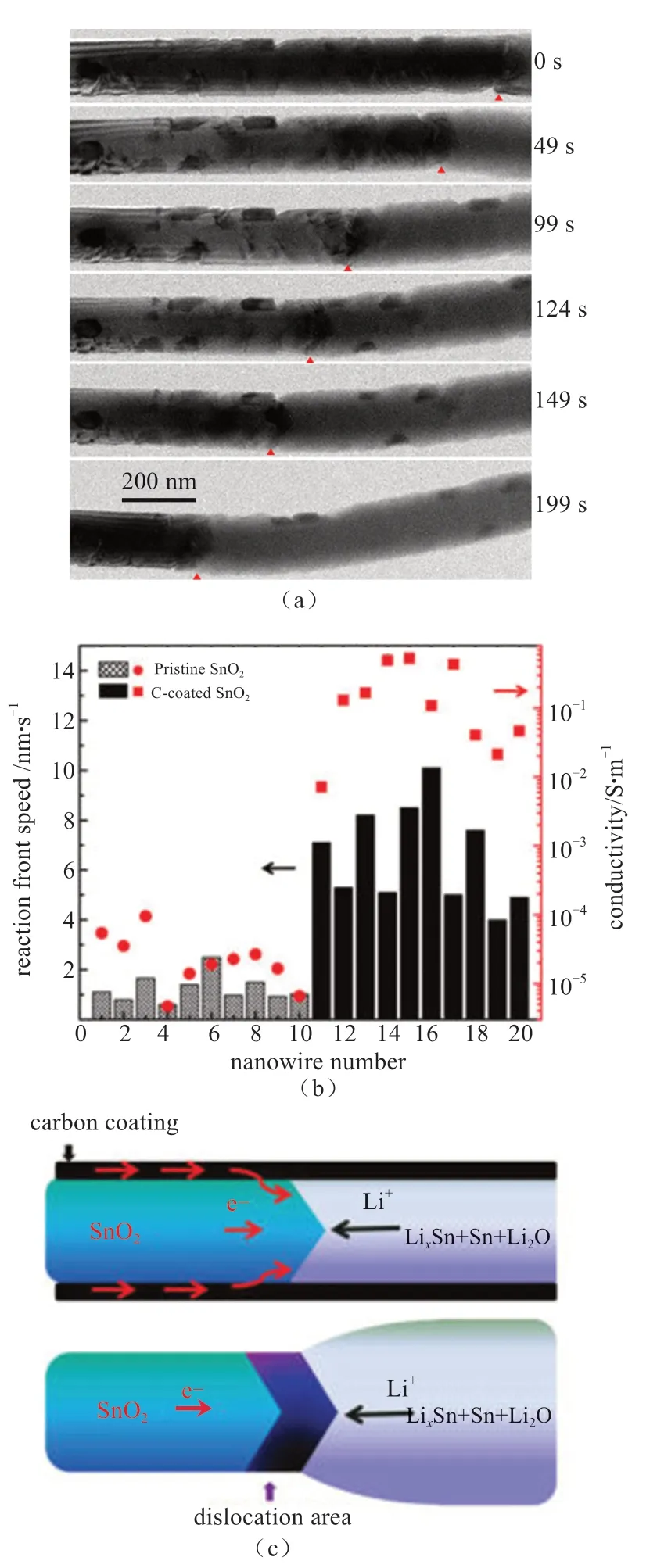
图7 (a)碳包覆SnO2纳米线在嵌Li过程中仅发生伸长,沿着径向基本没有发生膨胀(红三角为反应前端);(b)碳包覆和原始SnO2纳米线充电速率和导电性比较统计图。碳包覆设计使SnO2纳米线的导电率提高3 ~4个数量级。碳包覆SnO2纳米线嵌Li速度也比原始的SnO2纳米线提升了5倍;(c)碳包覆与原始SnO2纳米线嵌Li机理示意图[38]Fig.7 (a)The carbon coated SnO2 nanowire elongated without radial expansion (the reaction front is marked by red triangles); (b) Statistics of the reaction front migration speeds and conductivity of the C-coated and pristine SnO2 nanowires.The conductivity of the C-coated nanowires was measured to be 3~4 orders of magnitude higher than that of the pristine SnO2 nanowires, combing with the lithiation rate was enhanced for 5 times after carbon coating; (c)Lithiation mechanisms of the carbon coated and pristine SnO2 nanowire [38]
Li-O2电池的负极通常为纯金属锂,然而,金属锂非常活泼,暴露于空气中易燃烧和爆炸,难以管理和运输。为此,YANG等[42]应用In situ TEM技术,在CO2气氛条件下运用电化学电镀法发明了一种制备空气稳定锂球(air stable lithium spheres,ASLSs)的新技术(图9)。实验装置设计与Li-O2电池相同,仅是ETEM环境中的气体变为CO2,随着反应的进行,在三相接触点会有一金属球的形成。这个ASLSs内部是金属锂,外壳为14~80 nm厚的Li与CO2之间的电化学反应产物Li2CO3。当放置在空气环境中时,ASLSs不与空气中的水等反应并可维持其稳定的核壳结构。ASLSs可以作为一个LIBs中的阳极,它们表现出和锂金属相同的电化学性质,表明其表面的Li2CO3层是良好的锂离子导体。ASLSs的空气稳定性归因于表面的Li2CO3层,它几乎不溶于水,在室温下与空气中的氧气和氮气均不反应,从而钝化内部锂核。这些结果表明对于下一代锂金属来说,相比于纯锂金属电极ASLSs更加安全和稳定。
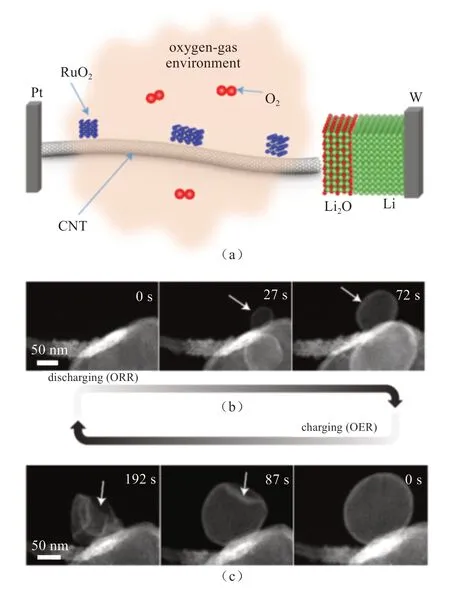
图8 (a)原位环境透射电镜中搭建Li-O2 金属空气电池结构示意图;(b)放电过程中微观形貌变化;(c)充电过程中微观形貌变化[41]Fig.8 (a) Schematic that illustrates the configuration of the Li-O2 nanobattery in an environmental TEM chamber; (b)The time resolved high-angle annular dark-field scanning TEM images depict the morphological evolution of the discharging product (ORR); (c) The images illustrate the morphological evolution on charging (OER) [41]
3.2 钠金属-空气电池

图9 (a)实验装置的示意图。Li 电镀装置由CNT 阴极,Li2O 固体电解质和Li 阳极组成;(b)在CO2 环境中生成ASLS 的过程图。在负电压条件下,在CNT、Li2O 和CO2的结合点处会产生一个金属锂球[42]Fig.9 (a) experimental setup.The Li plating device consists of a CNT cathode, a Li2O solid electrolyte, and a Li anode;(b) The generation of an ASLS in a CO2 environment.Upon applying a negative potential, a sphere emerged at the CNT,Li2O, and CO2 triple point.The sphere with a core-shell structure grew with time [42]
钠金属-空气电池由于其低成本与高理论能量密度而引起了大家的广泛关注。然而,其在实际应用中由于受到空气阴极中缓慢的氧还原反应(ORR)和析氧反应(OER)动力学引起的低循环性、低倍率性能和低充放电效率的困扰。因此,贵金属如Au、Pd、Pt、Ru等和过渡金属氧化物如MnO2、Co3O4、Fe2O3、CuO等被用作电催化剂以促进电池中缓慢的ORR和OER。然而有研究表明,相比Li-O2电池,Na-O2电池在没有催化剂的碳基电极作为空气阴极时亦可表现出良好的性能。在碳阴极的Na-O2电池中,人们观察到优于Li-O2电池的较低ORR和OER过电位以及更好的循环性,使得许多研究人员认为在基于液体非水电解质的Na-O2电池中不需要电催化剂,碳基材料本身就可以作为有效的电催化剂。然而,也有其他研究者使用贵金属和过渡金属氧化物作为Na-O2电池电催化剂的报道。但是,Na-O2电池中是否需要电催化剂?如果是,则在Na-O2电池中是如何进行电催化过程的,包括催化剂的结构演变以及放电产物的成核和生长。最近球差校正环境透射电子显微镜(ETEM)的出现使金属-空气电池的原位研究成为现实,这为揭示Na-O2电池中的关键问题指明了方向。
为了深入阐明催化剂在金属空气电池中的电催化作用,LIU等[43]设计了一种全固态Na-O2纳米电池,分别以原始MnO2纳米线和表面沉积Au纳米颗粒的MnO2纳米线为空气阴极,以自然形成的Na2O为电解质和金属Na在O2环境组装成一纳米电池[图10(a)]。Na-O2电池的原位ETEM研究揭示Au纳米颗粒具有明显的ORR催化活性,Au/MnO2纳米线表面形成NaO2可导致体积增加18倍[图10(b~i)],Au催化剂的添加显著提高了材料的催化反应活性;而没有Au沉积的MnO2纳米线则反应缓慢,没有超氧化物的形成。
3.3 钾金属-空气电池
地壳中的K元素含量比Li高出约880倍,K的价格远低于Li[44]。 且与Na相比,K/K+的标准电位[2.92 V(νs.SHE)]a/Na+[-2.71 V(νs.SHE)]意味着K阳极基电池的工作电压可以更高[45]。另外,因为K+的路易斯酸性比Li+和Na+弱,所以它在电解质和界面中具有更快的迁移速率,有助于电池表现出更好的性能。虽然钾电池在未来显示出巨大的潜力,但钾电池的微观反应机理尚不清楚。
以金属钾-二氧化碳电池为例,目前尚不清楚其准确的充放电反应方程式。为此,ZHANG等[46]使用ETEM,在CO2气氛下,将金属K与CNT组装成一纳米电池,原位研究了K-CO2电池在充放电过程中的微观结构变化(图11)。在对CNT施加负偏压时,在CNT、K2O的接触点处会有一纳米球形成。该球为空心结构,且随着反应的进行逐渐长大。反应进行22 s后,球可以长到174 nm。在生长的过程中,球有时会将CNT推离接触点但仍会继续生长,表明生成的球可以传输电子和离子。当施加反向电压时,球随着气体的排出逐渐缩小,最终在16 s后完全分解。再次对CNT施加负偏压时,一个新球再次出现在同一个位置;再次施加正压,球又发生收缩,表明该电池可以反复充放电。通过进一步的电子衍射(EDP)及电子能量损失谱(EELS)发现CNT上放电产物为K2CO3和CO。CO的释放导致了纳米球的形成,其中K2CO3为外壳,内部为中空结构。施加正压时(充电反应)K2CO3还会进行分解,完成电池的反复充放电过程,纳米球的形成方式与离子扩散和电子传输路径密切相关。通过原位电镜表征,我们掌握了K-CO2电池在充放电过程的微观变化,这对于理解电池的工作原理和探索其稳定性具有重要意义。
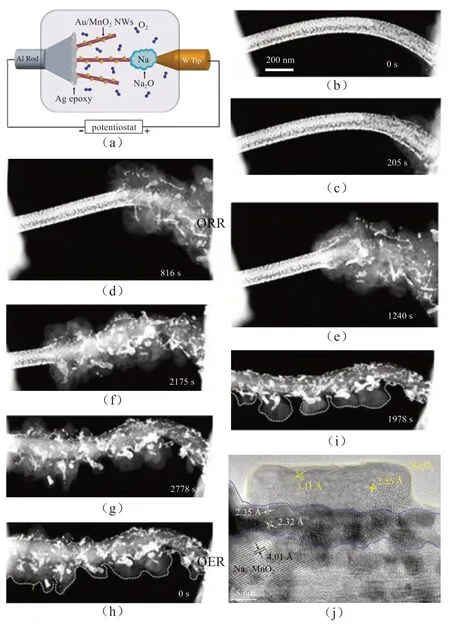
图10 (a)实验装置的示意图。该电池由Au/MnO2 纳米线和O2阴极,Na2O电解质和Na阳极组成。(b~g)ORR期间NaO2放电产物的结构演变。在施加负电位时,在Au/MnO2纳米线和Na2O接触处形成气泡状NaO2(b,c),然后形核长大并沿纳米线迅速传播,导致体积增加18倍(d~e)。由于NaO2的歧化反应生成物为Na2O2和O2(f~g),O2的释放使纳米线又发生收缩。(h~i)Au/MnO2纳米线的充电反应过程;(j)充电反应后产物的HRTEM图像[43]Fig.10 (a) Schematic of the experimental setup.The battery consists of a Au/MnO2 NW and O2 cathode, a Na2O electrolyte and a Na anode; (b~g) Structure evolution of the NaO2 discharge product during ORR.Upon applying a negative potential to the Au/MnO2 NW, bubble-like NaO2 nucleated on the right contact where the Au/MnO2 NW and Na2O intersects (b~c), which then propagated along the NW,causing an 18 times volume increase (d~e).The discharge product shrank as a result of the disproportionation of NaO2 to Na2O2 and O2 (f~g); (h~i) Charge process of the Au/MnO2 NW.(j) HRTEM image of the residue discharge products after the charging process [43]
4 总结与展望
In situ TEM技术作为一项探究电化学反应的重要技术,具有超高的空间和时间分辨率,可在原子尺度上对许多电池的电化学反应过程进行原位表征。此技术在表征电池电化学反应研究方面具有显著优势:①可达到原子尺度的高空间分辨率;②动态观察整个反应过程;③完整的结构和化学成分表征;④可原位操作。
虽然,In situ TEM技术在研究电化学问题方面很有前途,但是其与宏观电池的反应条件还略有不同,还有巨大的改善空间:①通过控制电流控制电池的充放电,使其与宏观电池充放电过程相对应;②研究阳极材料和SEI层在电化学反应过程中的精细变化;③在平台上配合更多的表征和操作工具以便探查出更多的样品信息;④该技术扩展到其他电化学研究领域。我们相信发展In situ TEM表征技术可以更好地解释电池在使用过程中所遇到的关健问题,推进高能量密度、高功率密度和长循环寿命的先进电池的发展,这对于电动汽车电池和风能、太阳能等发展也至关重要。
在未来研究中,亦可以应用In situ TEM技术结合冷冻电镜关注LIBs中的锂金属枝晶的生长及SEI层的形成过程[47-48]。由于锂金属活性强,在空气和电子辐射下都不太稳定,传统的TEM技术很难用于研究其精细结构。在传统TEM电子束的照射下,锂枝晶的形貌和结构会被严重破坏。为解决此问题,CUI等[47]参考生物冷冻电镜样品的制备方法,先在铜网上采用电化学方法沉积金属锂枝晶,然后清洗掉其表面残余的液体电解质,随后再迅速将其置于液氮之中用于冷冻电镜观察。此方法制备的锂枝晶样品表面干净,且在电子辐射下相当稳定,照射10分钟也不会被破坏。此外,KOURKOUTIS等[48]应用类似的冷冻电镜技术对SEI膜的结构和成分也进这行了详细的表征。

图11 放电状态和部分充电状态产物的EDP 和EELS 表征。(a)原始CNT 与K2O 接触前形貌;(b)在第一次放电过程中产生球,衬度及线扫描结果(红色轮廓)表明球为中空结构。第一次充电后,球缩小(c)并最终消失(d);原始CNT(e);处于放电状态的球(f);处于部分充电状态的球(g)和处于完全充电状态的产物(h)的EDP;(i ~j)分别是来自放电(b)和部分充电的球(c)的low loss 和core loss 的EELS 谱。红色和蓝色的谱图分别是从第一次充电和放电过程中获得的[46]Fig.11 Structure and phase identifications by electron diffraction patterns (EDPs) and electron energy loss spectroscopy(EELS) of the discharged and partially charged products.(a) The morphology of the pristine CNT contacting K2O; (b) A ball was generated during the first (1st) discharging process, the line scan result (red profile) indicates the hollow structure of the ball.The ball shrank (c) and finally disappeared after the 1st charging (d); (e ~h) EDPs of the pristine CNT (e), the ball at the discharged state (f), the ball at partially charged state (g), and the products at the completely charged state (h); (i ~j)are low-loss and core-loss EELS spectra from the discharged (b) and partially charged ball (c), respectively; The detection area is only one point marked by blue; The Red and blue profiles were acquired from the 1st charge and discharge processes,respectively [46]
近期,三维重构、integrated Differential Phase Contrast(iDPC)、HADDF-STEM等新技术和新方法亦发展迅速[49-51],若能配合使用In situ TEM技术,将对电池的微观机理研究产生更为重要的影响。In situ TEM技术的发展对于电池材料乃至整个材料科学发展来说既是机遇也挑战,是大有可为的。
猜你喜欢
幼儿100(2024年19期)2024-05-29 07:43:34
新能源汽车供能技术(2021年1期)2021-10-14 08:59:48
原子与分子物理学报(2021年2期)2021-03-29 07:31:10
电子制作(2019年23期)2019-02-23 13:21:36
天然产物研究与开发(2018年5期)2018-06-13 03:23:54
材料科学与工程学报(2016年4期)2017-01-15 13:35:33
材料科学与工程学报(2016年2期)2017-01-15 13:34:35
电源技术(2015年5期)2015-08-22 11:18:02
电测与仪表(2014年17期)2014-04-04 11:57:00
河南科技(2014年12期)2014-02-27 14:10:29
