
聚苯乙烯包覆石蜡相变微胶囊的制备及性能分析
2019-11-18 10:21:32李凤艳胡荣荣徐远航赵天波
石油化工高等学校学报 2019年5期
李凤艳,胡荣荣,徐远航,赵天波
(1.北京石油化工学院化学工程学院,北京102617;2.北京理工大学化学与化工学院,北京100081)
自20世纪70年代至今,相变微胶囊已应用于节能建材[1]、热能传递、太阳能利用[2]、储热调温[3-5]等领域,成为国内外研究的热点。目前,制备相变材料微胶囊所用壁材的种类有很多,常采用原位聚合法制备壁材为蜜胺树脂和脲醛树脂类微胶囊,但受其制备工艺及材料内在性质的影响,在胶囊壁中可能残留甲醛,因而限制了其应用[6-7]。因此,研制无甲醛壁材的相变微胶囊成为目前研究的热点。聚苯乙烯材料由于具有质轻、绝缘、绝热、防震、隔音、防腐蚀、抗水、化学性质稳定等优良性能,被广泛地应用在建筑、轻工、纺织、航运和国防等领域[8-9]。聚苯乙烯(PS)源自于石油产品,其生产过程中的排污较容易达到排放标准,它的制品加工过程更加符合环境保护要求,无污染物产生,为洁净生产。
目前已发表的文献中,制备聚苯乙烯微胶囊往往选择硬脂酸丁酯[10-11]和正十八烷[12-13]为芯材,而硬脂酸丁酯的相变点为20℃,相变焓为140 J/g;正十八烷的相变点为28℃,相变焓为240 J/g,但价格昂贵以及难以实现工业化制约了这类相变材料的应用[14]。石蜡作为相变材料,因具有廉价易得、安全无毒、密度小、相变潜热大、过冷和不析出等优点,可以作为制备相变微胶囊的芯材。黄云峰等[15]采用乳液聚合法制备了以聚苯乙烯为壁材,正十四醇为芯材的MA/PS微胶囊,其中MA/PS微胶囊的熔化焓和结晶焓分别为162.1、124.9 J/g,正十四醇的质量分数为73.7%。Y.Ming等[16]采用类悬浮聚合法合成了苯乙烯-二乙烯基苯(DVB)共聚物壳包覆正十八烷的相变微胶囊,微胶囊的平均直径约为80 μm,热焓约为126 J/g。黄全国等[17]采用类悬浮聚合法制备了聚苯乙烯/石蜡相变微胶囊,石蜡质量分数为20%~50%的微胶囊壁材对芯材保护效果较好,其中石蜡质量分数为50%的微胶囊对应相变潜热值最高达到92.4 J/g;当石蜡质量分数提高到60%时,微胶囊的形貌会变得不规则。
为满足石蜡微胶囊代替石蜡作为PBX炸药体系的钝感剂起到较好的钝感效果,期望得到芯材含量较高的微胶囊,同时也解决了石蜡高温易融化、易迁移和分布不均匀等问题。本研究采用悬浮聚合法制备以聚苯乙烯为壁材、石蜡为芯材的相变材料微胶囊,针对黄全国等[17]制备PS壁材-石蜡相变微胶囊的实验方法进行了改进,考察乳化时间、聚合时间、交联剂配比以及芯壁比等工艺条件对相变微胶囊的化学结构、表观形貌、热性能和热稳定性的影响,从而提高芯材石蜡质量分数和热焓值,改善石蜡的热性能和热稳定性。
1 实验部分
1.1 实验仪器与试剂
试剂:苯乙烯(St,化学纯)、二乙烯基苯(DVB,纯度>80%)、聚乙烯吡咯烷酮(PVP,分析纯)、石蜡(Tm在58~60℃,工业级)、偶氮二异丁腈(AIBN,化学纯)、质量分数95%乙醇(工业级),均为市售。
仪器:DHG-9053A电热恒温鼓风干燥箱,上海一恒科技有限公司;Quanta-200扫描电子显微镜,捷克FEI公司;DSC-60差示扫描量热分析仪,岛津公司;TG-DTA 6200 LAB热重分析仪,岛津公司;BRUKER UECIOR 22型傅立叶变换红外光谱仪,布鲁克光谱仪器公司。
1.2 石蜡微胶囊的制备
称取一定量的乳化剂PVP和去离子水于500 mL三口烧瓶中,60℃下低速搅拌,制成水相;称取一定量的石蜡放入100 mL单口烧瓶中,60℃加热熔化后,依次加入单体和交联剂,最后加入引发剂AIBN粉末,并震荡使之溶解,配制油相;将油相倒入水相,在1 500 r/min下机械搅拌乳化10 min;然后,水浴升温至80℃,700 r/min机械搅拌下聚合反应5 h;将反应后得到的样品趁热抽滤,并用70℃的水和乙醇分别洗涤3次,以洗去微胶囊表面附着的微量石蜡、残留的分散剂和未反应的单体,最后将得到的产物放入50℃的烘箱内干燥至恒重。
1.3 石蜡微胶囊的表征
化学结构测定:采用傅立叶变换红外光谱仪测试纯石蜡、壁材聚苯乙烯(PS)和微胶囊的化学结构。
形貌测定:采用场发射扫描电子显微镜观察微胶囊的表面形貌。
热性能测试:采用差示扫描量热仪测试壁材聚苯乙烯(PS)、纯石蜡和微胶囊的热性能。利用式(1)计算微胶囊芯材石蜡的质量分数:

式中,ΔH微胶囊指制备的微胶囊样品的固-液相变潜热,J/g;ΔH纯石蜡指纯石蜡的固-液相变潜热,J/g。
热稳定性测试:采用热重分析仪测试壁材聚苯乙烯(PS)、纯石蜡和微胶囊的热稳定性能。
2 结果与讨论
2.1 微胶囊的红外光谱表征
对壁材聚苯乙烯(PS)、纯石蜡和相变微胶囊样品进行红外光谱测试,FTIR谱图如图1所示。
由图1中可以看到,在壁材PS的FTIR谱图中,在2 922、2 849 cm-1处的峰对应的是-CH2中C-H键的伸缩振动峰,1 492、1 451 cm-1处同时出现尖锐的中等强度吸收峰为苯环骨架振动吸收峰,在690、756 cm-1处出现的强吸收峰对应的是单取代苯环的弯曲振动吸收峰。在纯石蜡的FTIR谱图中,波长为2 849、2 918 cm-1处的吸收峰为脂肪链中C-H键伸缩振动吸收峰;在1 463 cm-1波长处出现的吸收峰为C-H弯曲振动吸收峰;波长为720 cm-1处的吸收峰是-CH2的面内摇摆吸收峰,表现为具有4个以上-CH2的长链烷烃。观察微胶囊的FTIR谱图可以发现,微胶囊的谱图中包含有石蜡和PS全部的特征吸收峰,没有出现新的特征吸收峰,说明石蜡和PS之间没有发生化学反应,微胶囊中石蜡没有发生任何变化,表明石蜡被成功包覆形成了微胶囊。

图1 红外光谱分析图谱Fig.1 FTIR spectra of three samples
2.2 乳化时间对微胶囊热性能的影响
以乳化时间分别为10、20、30 min制备石蜡相变微胶囊,利用差示扫描量热仪测试纯石蜡和微胶囊样品的热性能,测试的纯石蜡和微胶囊样品的热性能数据见表1。
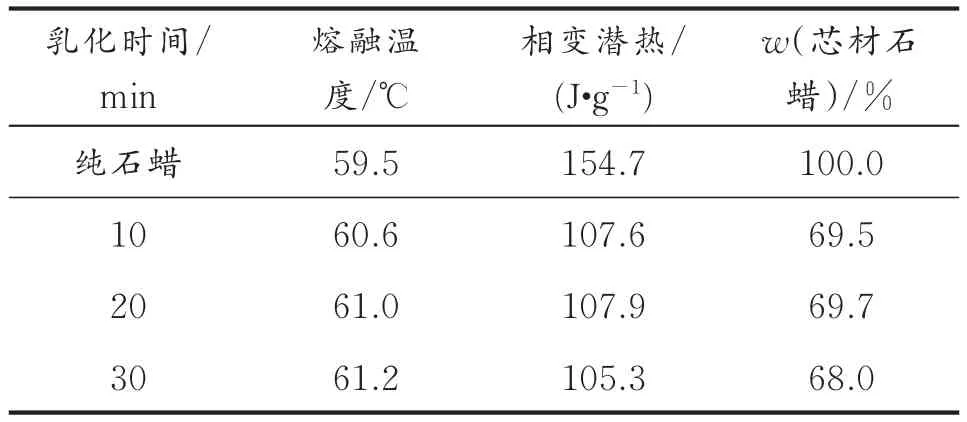
表1 不同乳化时间制备相变微胶囊的热性能Table1 Thermal properties of MicroPCMs prepared in different emulsifying times
从1表可以看出,乳化时间分别为10、20、30 min制备的微胶囊的相变始末温度比纯石蜡的相变始末温度均有推迟,这说明壁材对芯材石蜡具有一定的热阻滞作用。乳化时间对微胶囊的热性能影响不大,不同乳化时间制备的微胶囊的相变潜热和芯材石蜡质量分数都接近,以乳化时间分别为10、20、30 min制备微胶囊的相变潜热对应为107.6、107.9、105.3 J/g,芯材石蜡质量分数分别为69.5%、69.7%、68.0%。
2.3 交联剂DVB配比对微胶囊形貌的影响
采用单体St与交联剂DVB的质量比分别为5∶1、6∶1、7∶1、8∶1、9∶1制备了石蜡相变微胶囊,利用扫描电子显微镜观察表观形貌,其SEM照片如图2所示。

图2 不同交联剂DVB配比制备微胶囊的SEMFig.2 SEM micrographs of microcapsules with different contents of the crosslinking agent DVB
由图2可以看出,不同交联剂配比制备的微胶囊球形度较差且有明显的缩皱现象,其中以m(St)/m(DVB)为5∶1制备的微胶囊缩皱最为严重,以m(St)/m(DVB)为 7∶1、8∶1、9∶1制备的微胶囊表面黏附有许多碎屑且颗粒大小不均匀,而以m(St)/m(DVB)为6∶1制备的微胶囊形貌相对较好,缩皱较m(St)/m(DVB)为5∶1制备的微胶囊有所改善,表面碎屑比m(St)/m(DVB)为 7∶1、8∶1、9∶1制备的微胶囊较少。因此,选用m(St)/m(DVB)为6∶1来制备聚苯乙烯/石蜡相变微胶囊较为适宜。
2.4 聚合时间对微胶囊的性能的影响
图3为采用聚合时间分别为3、5、7 h制备微胶囊的SEM照片。

图3 不同聚合时间制备微胶囊的SEMFig.3 SEM micrographs of microcapsules with different polymerization times
由图3可以看到,聚合时间为3 h制备的微胶囊呈现球形,表面黏附有少许碎屑且有缩皱,颗粒大小相对均匀;聚合时间为5 h制备的微胶囊也呈现球形,表面黏附有许多碎屑,但颗粒大小不均匀;聚合时间为7 h制备的微胶囊有的呈球形,有的呈棒状,表面较光滑且颗粒大小较均一。
图4为聚合时间分别为3、5、7 h制备相变微胶囊的DSC曲线。

图4 不同聚合时间制备相变微胶囊的DSC曲线Fig.4 DSC curves of phase change microcapsules prepared in different polymerization times
由图4可以看到,壁材聚苯乙烯(PS)的DSC曲线在30~80℃没有出现吸热峰,这说明壁材聚苯乙烯在该温度区间没有发生相变。纯石蜡和微胶囊的DSC曲线在30~80℃出现两个吸热峰,其中在35~50℃出现的小吸热峰对应的是石蜡的固-固相变,在50~70℃出现的大吸热峰对应的是石蜡的固-液相变。不同聚合时间制备的微胶囊发生相变的温度区间比纯石蜡的相变温度区间较宽,芯材发生固-液相变的结束温度明显推迟,这说明壁材对于芯材的释放具有一定的阻滞作用和渗透性能。
表2是不同聚合时间制备相变微胶囊的热性能。
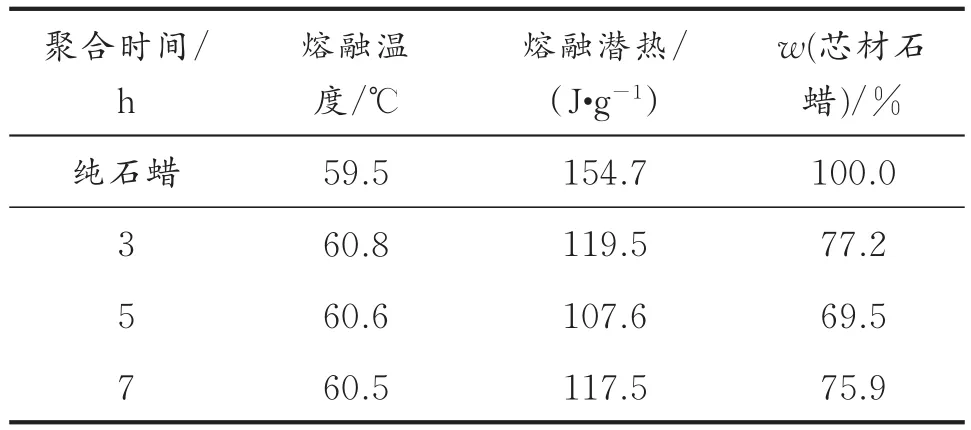
表2 不同聚合时间制备相变微胶囊的热性能Table2 Thermal properties of MicroPCMs with different polymerization times
由表2可以看出,不同聚合时间制备的微胶囊的相变熔融温度比纯石蜡的相变熔融温度均有小幅度升高,这说明壁材对芯材石蜡起到一定的热阻滞作用。聚合时间分别为3、5、7 h制备的微胶囊的相变潜热为 119.5、107.6、117.5 J/g,微胶囊中芯材石蜡的质量分数分别为77.2%、69.5%、75.9%。
2.5 芯壁比对微胶囊性能的影响
2.5.1 芯壁比对微胶囊形貌的影响 图5为采用芯壁比分别为 1∶1、2∶1、3∶1制备微胶囊的 SEM照片。
由图5可以看到,芯壁比为1∶1制备的微胶囊呈球形,表面光滑但有严重缩皱;芯壁比为2∶1制备的微胶囊呈球形,颗粒大小不一致且表面有明显缩皱;芯壁比为3∶1制备的微胶囊有些呈球形,有些呈棒状,且有许多碎屑存在。芯壁比为1∶1制备的微胶囊有严重缩皱,可能是由于芯材的加入量较少致使壁材包覆少量芯材后,随着聚合反应结束和温度下降至室温,使壁材发生收缩和芯材凝固而造成微胶囊有严重缩皱现象。芯壁比为3∶1制备的微胶囊缩皱有所减缓,但有许多碎屑存在,是因为芯材的加入量增加使壁材能够包覆足够多的石蜡芯材而减少了,由于石蜡凝固造成的缩皱,但石蜡过剩使油相液滴相互碰撞形成大液滴而致使壁材不能完全包覆,造成许多壁材和芯材碎屑存在。

图5 不同芯壁比制备微胶囊的SEMFig.5 SEM micrographs of microcapsules with different core/shell mass ratios
2.5.2 芯壁比对微胶囊热性能的影响 图6为芯壁比分别为 1∶1、2∶1、3∶1制备相变微胶囊的 DSC曲线。
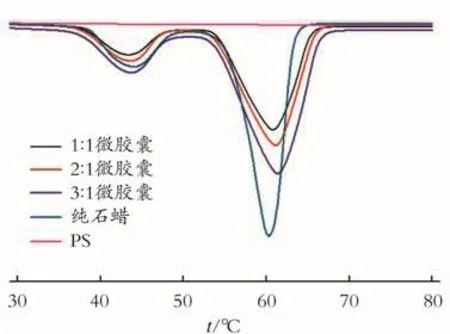
图6 不同芯壁比制备相变微胶囊的DSC曲线Fig.6 DSC curves of phase change microcapsuleswith different mass ratios of core and shell
根据图 6中芯壁比分别为1∶1、2∶1、3∶1制备相变微胶囊的DSC曲线,计算微胶囊的相变潜热和芯材质量分数。表3是不同芯壁比制备相变微胶囊的热性能。

表3 不同芯壁比制备相变微胶囊的热性能Table3 Thermal properties of MicroPCMs with different mass ratios of core and shell materials
由表3可以看到,不同芯壁比制备的微胶囊的相变温度比纯石蜡的相变温度都有所提升。随着芯壁比的增大,微胶囊的熔融潜热值逐渐增大,芯材石蜡质量分数也相继增大,这可能是因为随着芯壁比的增大,芯材石蜡的加入量增加,使壁材能够包覆足够的芯材形成微胶囊。
2.5.3 芯壁比对微胶囊热稳定性的影响 图7为芯壁比分别为 1∶1、2∶1、3∶1制备相变微胶囊的TGA曲线。
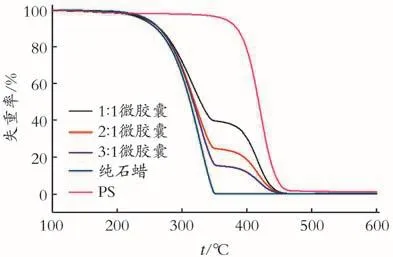
图7 不同芯壁比制备相变微胶囊的TGA曲线Fig.7 TGA curves of phase change microcapsuleswith different mass ratios of core and shell
由图7可以看到,石蜡的TGA曲线呈现一步分解过程,在170℃开始分解至350℃完全失重。壁材聚苯乙烯的TGA曲线也呈现一步分解过程,在270℃开始分解至475℃失重趋于平缓,但仍有1.3%的质量残留。微胶囊失重分两个阶段:第1阶段主要源于微胶囊中石蜡的挥发与分解;第2阶段主要是壁材的分解。由图7可以看出,芯壁比分别为1∶1、2∶1、3∶1制备的微胶囊由于石蜡的分解造成的质量损失分别为61.0%、75.7%、85.0%,并且芯壁比为3∶1制备的微胶囊中石蜡的分解温度有明显提升,同时在失重过程中可以观察到壁材对芯材的热阻滞作用。
3 结 论
采用悬浮聚合法制备了以聚苯乙烯为壁材、石蜡为芯材的相变微胶囊。结果表明,乳化时间对微胶囊的热性能影响不大;选用单体St与交联剂DVB的质量比为6∶1制备的微胶囊形貌较佳,缩皱和碎屑明显减少;聚合时间为3 h制备的微胶囊形貌较好,大小相对均一,相变潜热达到119.5 J/g,芯材石蜡质量分数为77.2%;而随着芯壁比的增大,微胶囊中芯材石蜡质量分数也相继增大,其中芯壁比为3∶1制备的微胶囊相变潜热增大到125.2 J/g,芯材石蜡质量分数高达80.9%,且热稳定性好。
猜你喜欢
天津科技(2022年7期)2022-07-29 08:42:48
现代食品(2022年10期)2022-06-08 05:55:26
上海第二工业大学学报(2021年4期)2022-01-22 07:11:34
天津科技(2021年7期)2021-07-29 13:47:06
天津化工(2021年1期)2021-01-05 16:42:05
林产工业(2020年8期)2020-08-31 06:58:26
天津科技(2020年7期)2020-07-31 09:10:56
食品安全导刊·下旬刊(2020年3期)2020-07-09 18:55:48
石油化工高等学校学报(2019年3期)2019-06-20 05:33:42
中国测试(2018年3期)2018-05-14 15:33:29
