
Ru改性碳纳米管催化甲醇分解的密度泛函理论研究
2019-11-15 05:47任瑞鹏
太原理工大学学报 2019年6期
任瑞鹏,李 云
(太原理工大学 煤科学与技术教育部与山西省共建重点实验室,太原 030024)
甲醇作为一种绿色可再生资源,被认为是传统燃料的合适替代品,它可以从天然气、煤炭和不同的可再生资源(如造纸黑液、动物粪便和生物质等)中获得[1-2]。甲醇的应用十分广泛:它可以分解制备氢气和一氧化碳,进而用作汽车和气体涡轮机的燃料;产生的氢气可以直接用于燃料电池[3];甲醇还可以用于直接甲醇燃料电池(DMFCs),而DMFCs被认为是21世纪最有潜力的电动汽车动力源[4]。了解甲醇的分解机理可以进一步控制甲醇分解路径并提高甲醇分解效率。
近年来,学者们研究了甲醇在不同金属催化剂上的分解机理[5-9]。JIANG et al[5]研究了在Pd(100)面上的甲醇完全脱氢反应,结果表明,C—H键的断裂对CH3OH和CH2OH更有利,O—H键的断裂对CHOH更有利。ZHOU et al[10]采用密度泛函理论广义梯度近似(DFT-GGA)方法研究了甲醇在Ni(100)表面的分解过程,并将Ni(100)与Ni(111)表面的分解过程做了详细的比较;结果表明,C—H键和O—H键的断裂均是Ni(100)上的有利反应路径,而在Ni(111)表面只有O—H键断裂是完善的反应路径。因此,甲醇分解反应可能是一种结构敏感的反应路径。CUI et al[11]采用重复平板模型和DFT-GGA方法研究了甲醇在Pt(111)、Pt缺陷、Pt-梯步面、Pt(110)(1×1)和Pt(110)(2×1)等5种Pt表面的分解机理;通过对甲醇在这些表面上分解反应机理的系统计算得出,甲醇在这些Pt基催化剂上的反应机理是相同的,即甲醇中O—H键发生断裂和甲氧基中C—H键发生断裂,最终产物都是H和CO.然而由于贵金属成本高、储量低,贵金属催化剂的大规模应用受到了限制,寻找其他成本低、效果好的甲醇分解催化剂成为这一领域的持续性热点。提高催化剂活性的一种方法是采用第二过渡金属对Pt表面进行改性。在各种Pt二元合金催化剂中,Pt因其较好的催化活性和CO耐受性常被用于甲醇氧化。JEMAL et al[12]将Pt负载在硼掺杂的石墨烯上,利用密度泛函理论研究了甲醇在该催化剂上的分解;结果表明,甲醇中的O—H键更易断裂形成CH3O作为初始步骤,且最终分解路径为CH3OH→CH3O→CH2O→CHO→CO.
单壁碳纳米管(SWNTs)可以看作是由单层石墨烯片层卷曲而成,具有独特的性质,如大的比表面积、中孔结构、耐高温性和特定的金属-载体相互作用。由于这些性质,碳纳米管被广泛用于不同的催化反应[13]。纯净的碳纳米管由于缺陷少,具有更高的均匀一致性,其催化活性相对较低。为了提高碳纳米管的催化活性,一般会用N、B、P或金属原子取代碳纳米管上的碳原子进行改性。本文选择Ru取代(6,0)碳纳米管上的一个碳原子构成新的催化剂(将其表示为Ru/CNTs),利用密度泛函理论研究了甲醇在Ru/CNTs表面的分解。
1 计算方法与模型
为了探讨Ru改性的单壁碳纳米管对甲醇分解的催化反应机理,构建了直径为0.470 nm的锯齿形(6,0)碳纳米管。该碳纳米管含有48个碳原子。采用Ru原子取代碳纳米管上的一个C原子,优化后的构型如图1所示。图1中,纳米管表面的5种不同吸附位分别为TopRu(T1)、TopC(T2)、bridgeRu-C(br1)、bridgeC-C(br2)和hollow.在所有计算中采用周期性模型,同时选择1.5 nm的真空度以避免相邻碳纳米管之间的相互作用,运用VASP(Vienna ab-initio simulation package)进行密度泛函理论计算[14]。构型优化过程中所有原子处于弛豫状态且不考虑自旋极化。应用具有广义梯度近似(GGA)[3]的Predew-Wang-91(PW91)函数[15]来计算相关能量,截断能设置为400 eV,布里渊区k点设置为5×1×1.采用NEB[16]方法搜寻甲醇分解中涉及的所有反应的过渡态(TS).
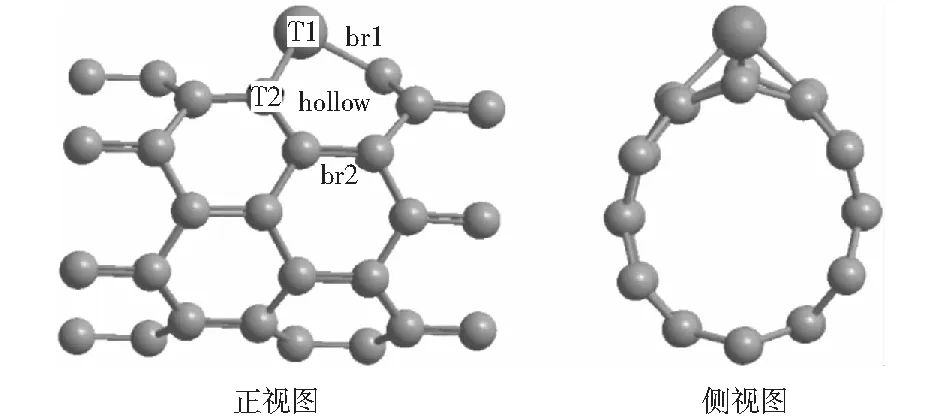
图1 Ru/CNTs的最优构型Fig.1 Front view and side view of Ru/CNTs optimal configuration
吸附能Eads为吸附前后各物质总能量的变化,它表示发生吸附的可能性和吸附的强弱程度。吸附能计算公式如下:
Eads=Eadsorbate/substrate-Esubstrate-Eadsorbate.
(1)
式中:Eadsorbate/substrate,Esubstrate,Eadsorbate分别表示吸附物在催化剂稳定吸附构型的总能量、催化剂稳定构型的能量和自由态分子的能量。
反应速率常数计算公式如下:
(2)
式中:h为普朗克常数;kB为玻尔兹曼常数;Ea为活化能;A为指前因子;T为温度。QIS和QTS分别代表初始态和过渡态的配分函数,计算公式如下:
(3)
式中:υi表示振动频率。
2 结果与讨论
2.1 甲醇及相关物种的吸附
本文通过计算甲醇和中间产物在催化剂表面的吸附能,确定了各物种的稳定吸附位(表1)和最终的吸附构型(图2)。优化后的构型将作为下一步分解的初始构型。

表1 甲醇及相关物种的吸附能和稳定吸附位Table 1 Adsorption energies and stable sites of CH3OH and related species in methanol decomposition

图2 甲醇及相关物种在最优吸附位的稳定结构Fig.2 The stable structures of methanol and related species at the optimal adsorption site
2.1.1甲醇的吸附
通过计算得出,CH3OH分子模型中C—H键长为0.110 nm,C—O键长为0.143 nm,O—H键长为0.097 nm;这与先前报道的理论值相吻合[17],说明所有计算的构型是合理的。为了确定甲醇在Ru/CNTs上的最佳吸附位,计算了5种可能存在初始结构的能量,即甲醇在5个不同位点的吸附。结果显示,甲醇分子中的氧原子最接近碳纳米管,C—O键与管轴方向平行,且在Ru/CNTs T1位点上吸附最稳定。可能的原因是:甲醇分子的HOMO是n轨道,处于全充满状态;LUMO是σ*轨道,能量较高,无法从表面接受电子;甲醇分子中O的p轨道与底物的Ru原子的d轨道不相匹配,相互成键能力弱。因此,甲醇在Ru/CNTs表面为弱的化学吸附[18]。吸附后甲醇中氧原子与表面之间的距离为0.232 nm,其吸附能为-0.53 eV;而甲醇在Ru(0001)[8]面上的吸附能仅为-0.38 eV.
2.1.2CHxO(x=1~3)的吸附
甲氧基(CH3O)稳定地吸附在T1位点;O原子与Ru原子相结合,O—Ru键长为0.192 nm,其吸附能为-3.32 eV;由于脱去了一个H原子,其吸附能远远大于CH3OH的吸附能。与甲氧基相同,CH2O也更倾向于吸附在T1位点;O原子与Ru/CNTs表面之间的距离为0.213 nm;C—O键与管轴方向平行,吸附能为-0.67 eV.同样,CHO优先吸附在T1位点上;CHO的C原子与Ru原子相结合,C—Ru键长为0.194 nm;吸附能为-1.67 eV.
2.1.3CHxOH(x=0~2)的吸附
CH2OH分子是甲醇分解过程中的重要中间体,其最优吸附位点为T1;C原子与Ru原子结合,C—Ru键长为0.209 nm,吸附能为-2.58 eV.CHOH具有与CH2OH类似的几何结构,稳定地吸附于T1位点;C原子与Ru原子结合,C—Ru键长为0.201 nm,吸附能为-2.30 eV.COH分子则更倾向于通过C原子吸附在br1位点;形成的C—C和C—Ru键长分别为0.143 nm和0.186 nm;吸附能为-3.00 eV.
2.1.4CHx(x=1~3)的吸附
CH3OH中C—O键断裂后形成的CH3稳定吸附于T1位;其C原子与Ru原子结合,C—Ru键长为0.211 nm,吸附能为-2.62 eV.与CH3相同,CH2吸附在T1位;C原子与Ru原子键合,C—Ru键长为0.194 nm,吸附能为-3.42 eV.CH更容易吸附在br1位点;CH中的C原子分别与Ru/CNTs中的Ru和相邻的C原子键合,形成的C—C和C—Ru键长分别为0.141 nm和0.187 nm,吸附能为-5.60 eV.
2.2 反应历程探索
为了探究甲醇在Ru/CNTs表面的分解机理,计算了图3中所包含的甲醇分解的所有可能的反应路径的活化能和反应热(表2)以及键长变化(表3).甲醇分解机理可能的反应路径主要有3条:路径1主要包括R1、R4、R5和R6;路径2包括R2、R7、R8、R9、R10、R11、R12、R13和R14;路径3包括R3、R15、R16、R17、R18、R19和R20.

图3 甲醇分解的完整路径图Fig.3 Complete path of methanol decomposition
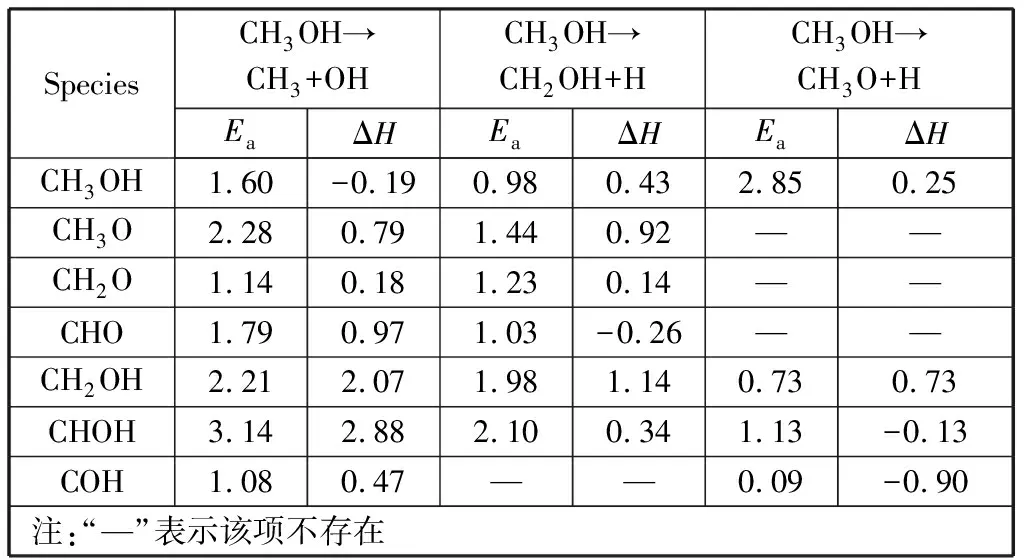

表3 甲醇及中间产物分解时各键键长的变化Table 3 Variation of bond length in the decomposition of methanol and intermediate products nm
2.2.1甲醇的分解
甲醇作为最简单的醇类,其分解可以通过C—O,C—H或O—H键断裂进行,以产生相应的中间体。因此,甲醇初始分解有以下3种不同的路径(R1,R2和R3).
1) R1:CH3OH→CH3+OH.CH3OH分子吸附在顶位作为反应物的初始构型。C—O键断裂形成CH3和OH;其中,CH3移动到相邻的T2位,OH吸附于T1位。此过程需要的反应热和活化能分别为-0.19 eV和1.60 eV.
2) R2:CH3OH→CH2OH+H.CH3OH分子中C—H键断裂形成CH2OH和H原子。此时CH2OH分子中C原子与Ru原子结合吸附于T1位,H原子移动到相邻的T2位。该反应的反应热和活化能分别为0.43 eV和0.98 eV.
3) R3:CH3OH→CH3O+H.CH3OH分子在O—H键断裂的情况下生成CH3O和H原子。该过程的反应热和活化能分别为0.25 eV和2.85 eV.如此高的活化能表明甲醇在Ru/CNTs表面初步分解的过程中O—H键不易断裂。
以上3种反应过程的初始态(IS)、过渡态(TS)及终态(FS)的构型如图4所示。通过3种路径活化能的对比,可以看出在甲醇分解过程中,C—H键最易发生断裂,而C—O键和O—H键的断裂不易发生。各反应的速率常数对比也验证了这一结果,如表4所示。
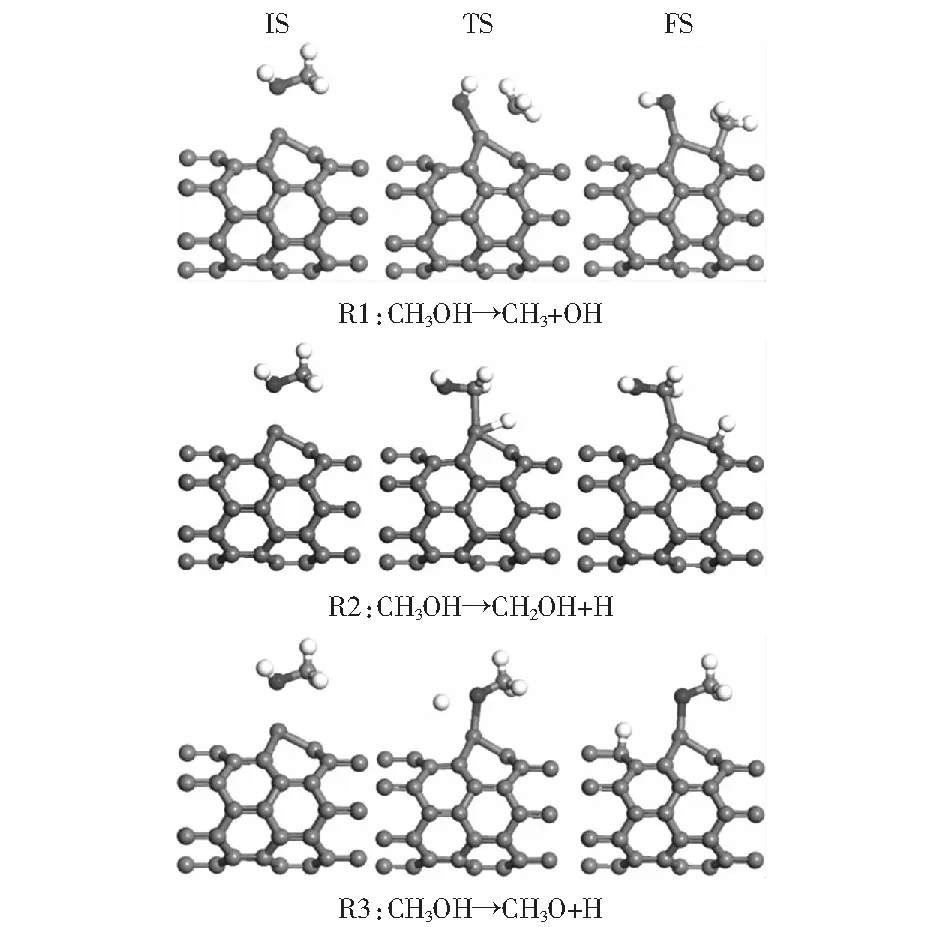
图4 甲醇在Ru/CNTs表面初步分解的初始态、过渡态及终态的结构Fig.4 IS,TS and FS strctures involved in the initial decomposition of methanol on Ru/CNTs
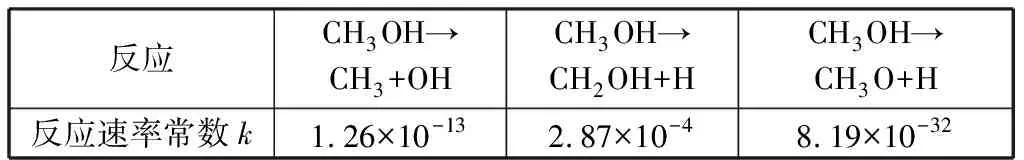
s-1
2.2.2CHx(x=1~3)的分解
由甲醇C—O键断裂生成的甲基CH3将进一步分解,即R4、R5和R6.各反应的结构如图5所示。
1) R4:CH3→CH2+H. 初始态时CH3稳定地吸附在T1位;经过C—H键的断裂生成CH2和H原子;在终态,CH2吸附于br1位,H原子则吸附于T1位。该过程的反应热和活化能分别为0.30 eV和0.76 eV.

图5 CH3分解各基元反应的初始态、过渡态及终态的稳定结构Fig.5 IS,TS and FS strctures involved in the CH3 decomposition
2) R5:CH2→CH+H.R4过程产生的CH2在T1位稳定吸附后继续分解产生CH和H原子,这一反应的反应热和活化能分别为0.63 eV和1.73 eV.该反应过程的活化能较大,说明此反应不易发生。
3) R6:CH→C+H.由R5产生的CH稳定吸附在br1位,分解后产生C原子和H原子。该过程的反应热和活化能分别为0.92 eV和1.19 eV.
2.2.3CHxOH(x=0~2)的分解
甲醇通过C—H键断裂形成的CH2OH进一步分解时,存在C—O、O—H和C—H键的断裂,即CH2OH的分解也存在3种不同的路径(R7、R8和R9).各基元反应的初始态、过渡态和终态如图6所示。

图6 CH2OH 分解各基元反应的初始态、过渡态及终态的稳定结构Fig.6 IS, TS and FS strctures involved in the CH2OH decomposition
1) R7:CH2OH→CH2+OH.CH2OH中C—O键断裂生成CH2和OH,CH2分子吸附于T1位,同时OH吸附于相邻的T2位作为终态。这一反应的反应热和活化能分别为2.07 eV 和2.21 eV,如此高的活化能表明CH2OH中的C—O键不易断裂。
2) R8:CH2OH→CH2O+H.CH2OH中O—H键断裂生成的CH2O分子通过C原子与Ru原子键合,H原子吸附于相邻的T2位。此过程所需的反应热和活化能分别为0.63 eV和0.73 eV.因此,相比于C—O键的断裂,该过程更容易发生,产生的CH2O继续分解。
3) R9:CH2OH→CHOH+H.CH2OH分子中C—H键断裂生成CHOH和H原子;CHOH通过C原子与Ru原子成键,吸附于T1位,H原子吸附于T2位作为终态。此反应的反应热和活化能分别为1.14 eV和1.98 eV,如此高的活化能表明该过程不易发生。为了了解该过程其他产物的分解情况,做了进一步计算。结果显示,CHOH分子中O—H(R10)、C—O(R11)和C—H(R12)键断裂的活化能分别为1.13 eV、3.14 eV和2.10 eV,表明CHOH分子中O—H键更容易断裂生成CHO分子。此外,COH因O—H键很容易断裂(R13)而形成CO和H原子,该过程的活化能仅为0.09 eV;而C—O键断裂(R14)则需要1.08 eV的活化能。
2.2.4CHxO(x=1~3)的分解
根据甲醇初始分解结果可知,生成CH3O的过程很难发生。但是为了系统地讨论甲醇分解机理,仍然对CH3O的分解进行了研究。计算结果表明,在Ru/CNTs表面甲氧基中的C—H键更易断裂(R15);因为此过程的活化能为1.44 eV,而C—O键断裂(R18)则需要2.28 eV的能垒。对于CH2O,其中的C—H(R16)和C—O(R19)键的断裂存在较强的竞争,两者的活化能分别为1.23 eV 和1.14 eV. CHO则是更容易出现C—H键断裂(R20,Ea为1.03 eV)而不是C—O键断裂(R17,Ea为1.79 eV).CH3O分解的所有基元反应的结构如图7所示。

图7 CH3O分解各基元反应的初始态、过渡态及终态的稳定结构Fig.7 IS,TS and FS strctures involved in the CH3O decomposition
3 讨论部分
3.1 态密度(DOS)分析
为了探究甲醇分子与Ru/CNTs之间的电子交互作用,本文对甲醇吸附前后的Ru/CNTs的态密度(density of states,DOS)做了详细计算。图8(a)是Ru/CNTs上吸附甲醇分子前后Ru原子的d轨道分布及d带中心的变化。可以看出,吸附甲醇分子后Ru原子的d轨道发生右移,且d带中心由原来的-1.75 eV变到-1.93 eV;这说明CH3OH与Ru原子之间发生了电子转移,使得CH3OH在其表面得到活化。图8(b)为Ru/CNTs上吸附甲醇分子后,甲醇分子及Ru原子的电子轨道分布。从图8(b)中可以看出,O原子的p轨道和Ru原子的d轨道重叠,说明甲醇与Ru原子之间的相互作用促使甲醇活化。

图8 甲醇吸附前后Ru/CNTs的态密度分布Fig.8 DOS distribution of Ru/CNTs before and after methanol adsorption
3.2 虚频的计算
为了验证各反应过渡态的准确性和计算反应速率,本文详细计算了所有反应的过渡态虚频,见表5.表5中有且只有一个虚频,充分说明所有过渡态数据真实有效,而且为反应速率的计算提供了数据。
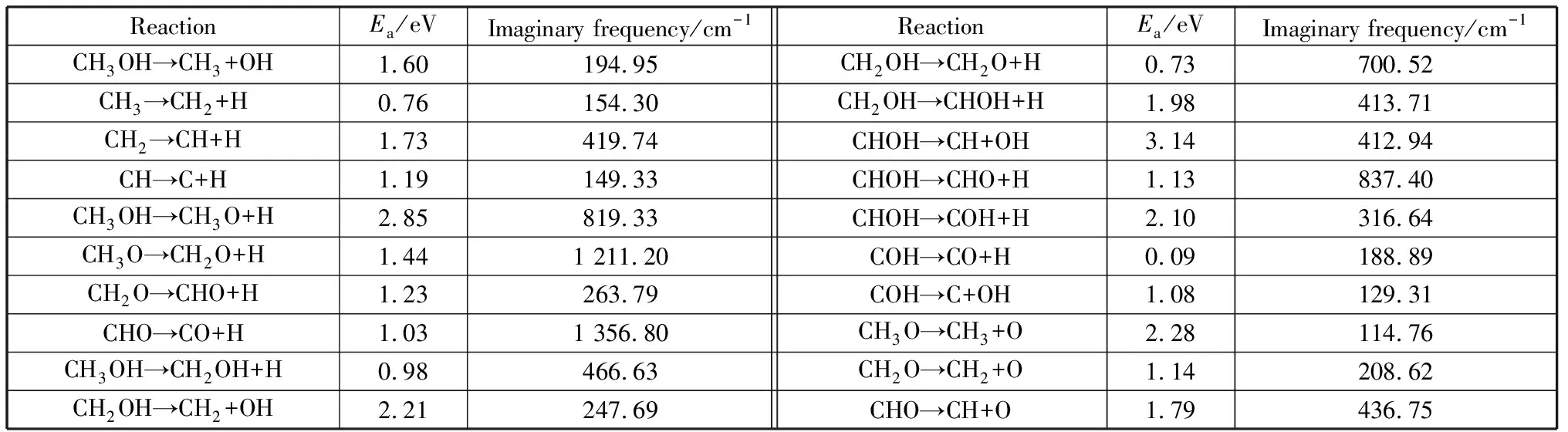
表5 甲醇分解中各基元反应的活化能及其对应虚频Table 5 The Reaction barriers and correspond imaginary frequency of each elementary reactions in methanol decomposition
4 结论
本文基于密度泛函理论,运用VASP软件和周期性模型计算了甲醇在Ru/CNTs表面分解的吸附能、活化能和反应速率,系统地讨论了甲醇在Ru/CNTs表面的分解机理。从计算结果可以得出如下结论:
1) 由甲醇及相关中间产物的吸附能可知,CH3OH、CH3O、CH2O、CHO、CH2OH、CHOH、CH3、CH2及CO分子的最佳吸附位为T1位,而COH、CH及C原子的最稳定吸附位是br1位。
2) 甲醇分解的第一步基元反应存在3条可能反应路径,即CH3OH→CH3+OH、CH3OH→CH2OH+H和CH3OH→CH3O+H.活化能和反应速率数据显示CH3OH→CH2OH+H反应最易发生,而CH3OH→CH3+OH和CH3OH→CH3O+H反应不易发生,即主反应路径为CH3OH→CH2OH→CHOH→COH→CO+H。
因此,Ru/CNTs是甲醇分解的有效催化剂,甲醇在其表面上分解的主产物是CO和H.本研究为制备新型高效的催化剂提供了线索。
猜你喜欢
四川大学学报(自然科学版)(2022年4期)2022-07-22
北京航空航天大学学报(2022年5期)2022-06-06
原子与分子物理学报(2022年3期)2022-03-05
化工设计通讯(2021年5期)2021-05-26
浙江农林大学学报(2020年2期)2020-04-22
物理化学学报(2020年1期)2020-04-02
青岛大学学报(工程技术版)(2019年2期)2019-09-10
电脑知识与技术(2018年3期)2018-03-21
哈尔滨理工大学学报(2017年1期)2017-04-08
科技视界(2016年24期)2016-10-11
