
成人散发型神经元核内包涵体病1例报告☆
2019-11-15 08:05:42伍瑶佳周娟冯刚华张文龙李海鹏
中国神经精神疾病杂志 2019年9期
伍瑶佳 周娟 冯刚华 张文龙李海鹏
神经元核内包涵体病(neuronal endouclear inclusion disease,NIID),又称神经核内透明质包涵体病(neuronal intranuclear hyaline inclusion disease,NIHID)或核内包涵体病(intranuclear inclusion body disease,INIBD), 是一种进展缓慢且致死的神经退行性疾病,其病理学特征是在中枢神经系统、周围神经系统以及内脏器官的细胞中存在嗜酸性玻璃样核内包涵体[1-2]。NIID临床表现多样,主要包括锥体和锥体外系症状、认知功能减退、癫痫发作、小脑共济失调、周围神经病变和自主神经功能障碍等[2]。现报告1例以精神行为异常、认知功能减退、发作性脑病、卒中样发作为主要临床表现的成人散发型NIID患者,分析其临床表现、影像学改变及皮肤病理学特征,并复习相关文献,旨在提高对本病的认识。
1 临床资料
患者69岁,女,因“精神行为异常、认知功能下降2年余,左侧肢体乏力、神志障碍、发热2 d”于2018年8月入院。患者于2016年9月因受凉出现咳嗽,自行服用感冒药后症状缓解。2 d后患者突然出现胡言乱语,反应迟钝,近记忆力减退,无偏瘫、偏盲、共济失调等症状,于我科住院,予以免疫球蛋白、护脑、营养神经、改善循环、抗病毒、抗感染等对症治疗后,精神症状较前好转后出院。2017年1月,患者再次出现类似症状,表现为自言自语、幻觉,觉得身边有很多人,别人衣服上有奇怪的东西,自知力下降,并有当众脱衣裤行为,与人沟通喜怒无常,同时伴有视力及听力下降。2018年8月8日,患者在吃中饭时突然出现左侧肢体乏力,神志不清,被送往当医院住院治疗。住院期间有发热,体温39℃,予以对症治疗之后(具体不详),患者意识逐渐清晰,左侧肢体仍乏力,精神症状、记忆力下降均较前加重。进食困难,大小便不自知,为求进一步诊治转至我院。既往2009年有脑梗死病史,无后遗症。个人史、月经婚育史及家族史无特殊。
入院查体:体温:37℃,脉搏:68次/min,呼吸:20次/min,血压:188/88 mm Hg。双肺呼吸音粗,可闻及散在湿罗音,心脏、腹部体查无异常。神经系统体查:神志嗜睡,言语含糊,思维混乱,记忆力、定向力差,计算力下降,理解力下降,自知力下降,右侧肢体可见活动,左侧肢体肌力2级,肌张力正常,双侧指鼻、跟膝胫试验、闭目难立征、共济运动、感觉粗测均不能配合,余体查无异常。
2016年9月住院辅助检查:血常规、肝肾功能电解质、乙肝两对半、肌酸激酶及同工酶、凝血功能、血脂、甲功三项、甲状腺过氧化物酶抗体、血氨、血乳酸、术前抗体、EB病毒抗体、肾内科结缔组织病全套,血管炎全套,肿瘤全套、结核抗体等检查,结果均未见明显异常。腰穿脑脊液检查:压力100 mm H2O;常规、生化、免疫、三大染色及细胞学结果均未见明显异常。脑电图:①背景异常:左右对称,右侧波幅较左侧明显;②背景β、θ波均增多。
2017年住院辅助检查:腰穿脑脊液检查:压力180 mm H2O;常规、生化、细胞学结果未见明显异常。脑脊液病毒性脑炎、自身免疫性脑炎相关抗体阴性。脑电图:右侧慢活动增多。视觉诱发电位:双侧视觉通路传导异常改变,双侧眼VEP检查双侧OZP100波潜伏期延长、波幅减低,波形分化、重复性差。
2016年至2018年头颅影像学均示脑白质广泛异常信号,DWI上皮层下区高信号呈动态变化(见图1)。取左小腿外踝上约10 cm处皮肤组织完善皮肤活检,结果示细胞核内可见 P62、泛素抗体强阳性染色的包涵体(见图2)。
2016、2017年,治疗上均予以免疫球蛋白、抗病毒、护脑、改善循环、补充B族维生素等对症治疗。2018年,予以丁苯酞注射液、护脑、改善循环、补充B族维生素等对症治疗。患者症状均有所好转。
2 讨论
NIID是一种罕见的多系统慢性进展性神经变性疾病。1968年,LINDENBERG[3]首次报告了 1例28岁男性患者,于儿童期发病,表现为智能发育迟缓、精神行为异常、小脑性共济失调等症状,尸检发现了大脑神经元及内脏细胞中存在大量典型的嗜酸性核内包涵体。1980年,SUNG等[4]报告了1例症状相似的患者,尸检中发现了同样的包涵体。当时将这一类疾病命名为 “神经元核内透明质包涵体病(NIHID)”。
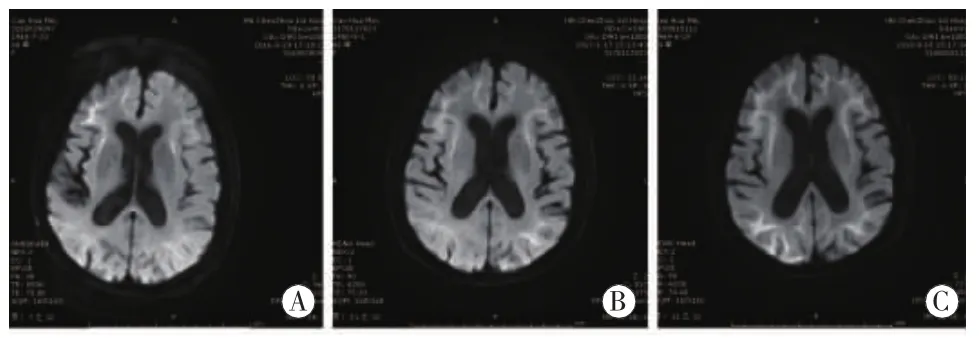
图1 影像学动态改变 A:2016年9月29日,DWI显示双侧额颞叶、外囊及右枕叶皮髓质交界处见斑条状高信号;B:2017年1月17日,DWI显示双侧额颞叶、外囊及双侧枕叶皮髓质交界处见斑条状高信号;C:2018年8月18日,DWI显示双侧额颞叶、外囊及双侧枕叶皮髓质交界处见斑条状高信号
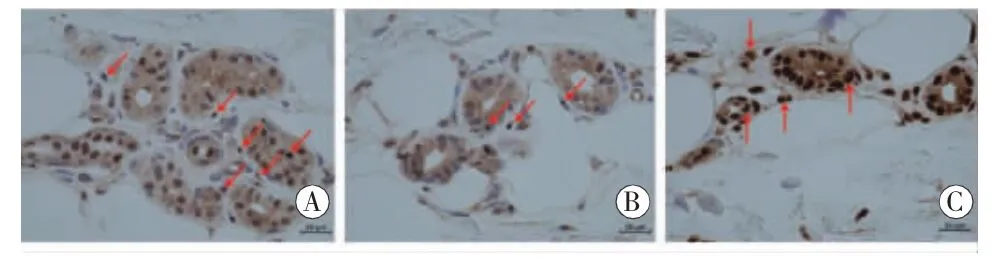
图2 免疫组织化学染色:A、B:部分汗腺细胞、脂肪细胞和纤维细胞的细胞核内可见圆形或类圆形包涵体,P62抗体染色阳性(箭头);C:泛素抗体染色强阳性的包涵体(箭头)
目前国内报告并不多,结合本例共为6例,均为女性,年龄60~74岁,其中1例有可疑家族史,余为散发型。临床表现为反复头晕发作 (1/6),反复发作性意识障碍(3/6),精神行为异常(3/6)、肢体无力(4/6)、消化道症状(4/6)、认知功能障碍(5/6)。由此总结我国患者常为反复发作形式起病,并可能以认知功能障碍、恶心呕吐、肢体无力为主要表现,但这还需大样本研究来验证。
NIID可在任何年龄段发病,TAKAHASHI-FUJIGASAKI[5]将NIID分为婴儿型、青少年型和成人型,在成人型中又分为散发型及家族型。不同亚型临床特征各异,其中成人散发型NIID发病年龄常为51~76岁,94.7%的患者以痴呆为首发症状和主要临床表现,意识障碍占39.5%,肌无力占27%,异常行为占26.3%,另有21%的成人散发型NIID的患者表现出亚急性发作性脑病的特征症状:发烧、头痛、呕吐和意识障碍[1,5]。本例NIID患者为成人散发型,主要表现为痴呆、精神行为异常、意识障碍、发作性脑病等症状,是成人散发型NIID的常见表现。但在本例患者初诊时,我们曾疑诊为病毒性脑膜炎、自身免疫性脑炎。因此,NIID的高度异质性易导致漏诊误诊。
影像学检查在NIID的诊断中可能成为重要的突破口。NIID的特征性影像学表现为DWI上皮髓交界处(U型纤维)出现有高信号,这种征象随着疾病进展,病灶将不断地沿皮层向后延伸,不会向髓质深部延伸;在T2WI及FLAIR序列上显示广泛的脑白质高信号[6-7],影像科医生容易误认为急性脱髓鞘改变。陈为安等[8]命名为皮层下绸带征、尿布征,NIID另一特征性MRI影像,为小脑萎缩,小脑蚓部旁及小脑中脚可见高信号[9]。
NIID的病理特征为嗜酸性核内包涵体,其广泛存在于中枢神经系统神经元和星形胶质细胞,以及周围神经系统与内脏细胞。神经元包涵体呈圆形,直径1.5~10 μm,位于核仁附近,泛素化及P62均为阳性,电镜下显示为无膜结构的致密纤维物质[1]。早期的NIID病例多为通过尸检、直肠活检及腓肠神经活检进行确诊。然而,直肠活检有穿孔的风险,腓肠神经活检只适用于有感觉障碍的患者。直到2011年SONE等[9]报告了在皮肤活检的标本中发现了脂肪细胞、成纤维细胞和汗腺细胞内可见嗜酸性的核内包涵体,这些核内包涵体泛素化阳性,且电镜显示这些核内包涵体特征与神经元细胞中的相同[10]。从此确定了皮肤活检在NIID诊断的重要作用。
目前NIID无明确统一的诊断标准,依据目前文献资料,以下条件可作为诊断参考标准:①中枢神经+周围神经+自主神经受累3组症状;②皮层下绸带征、尿布征,影像学以DWI在皮髓质交界处持续性高信号为主;③皮肤活检显示嗜酸性核内包涵体;④排除其他类似有类似影像表现疾病。本病目前无特异性治疗,我们使用免疫球蛋白及丁苯酞等药物治疗,患者短期疗效可,长期疗效有待观察及进一步研究。
猜你喜欢
自然杂志(2021年6期)2021-12-23 08:24:46
第一财经(2019年6期)2019-06-25 19:26:10
现代装饰(2018年5期)2018-05-26 09:09:01
天津诗人(2017年2期)2017-11-29 01:24:34
兽医导刊(2016年6期)2016-05-17 03:50:31
西南医科大学学报(2016年4期)2016-01-03 01:26:28
电源技术(2015年5期)2015-08-22 11:18:38
弹箭与制导学报(2015年1期)2015-03-11 15:32:06
云南畜牧兽医(2015年4期)2015-02-28 21:26:11
当代畜禽养殖业(2014年12期)2014-02-27 08:00:10
