
3-酮脂酰辅酶A硫解酶家族的分子特征、调节与药物靶向治疗
2019-11-15 07:17:50鲁洪廷曹玉红金振晓周京军
中国体外循环杂志 2019年5期
鲁洪廷,曹玉红,金振晓,周京军
作者单位:710032西安,空军军医大学生理与病理生理学教研室[鲁洪廷(2016级本科生),周京军],西京医院儿科(曹玉红),西京医院心血管外科(金振晓)
3-酮脂酰辅酶A硫解酶(3-ketoacyl CoA thiolase,3-KAT)是一类在脂肪酸β-氧化裂解反应中扮演重要角色的酶,包括线粒体型和过氧化物酶体(peroxisome)型,二者分别由不同基因编码。在线粒体该酶催化中长链及短链脂肪酸彻底裂解为乙酰辅酶A;在过氧化物酶体其底物则为极长链脂肪酸(≥C22)、长链脂肪酸和支链脂肪酸,经过有限的β氧化循环,缩短碳链,之后产物进入线粒体继续氧化(图1)。3-KAT是脂肪酸β-氧化最后一步,其活性的高低直接影响心肌对底物脂肪酸与糖的选择性及氧的利用效率。另在秀丽隐杆线虫观察到,该酶具有抗衰老作用,是去乙酰化酶Sirt2.1抗衰老的关键下游分子[1]。3-KAT在机体中的重要性使得有必要多角度阐释其分子特征。
1 线粒体3-KAT
1.1 分子结构与功能特点 线粒体三功能蛋白(mitochondrial trifunctional protein, MTP)是由 α-和β-亚基形成具有三重功能的多聚体,其中β-单位具有3-KAT的活性,而α-亚单位兼有加水酶和脱氢酶特性。多数学者认为,三功能蛋白是由4个β-亚单位和4个α-亚单位组成的八聚体,完成脂肪酸β-氧化的最后三步,最终将长链脂肪酸降解,生成乙酰辅酶A。由于线粒体MTP的化学名称为羟烷基辅酶A脱氢酶,所以人线粒体3-KAT的编码基因也称羟烷基辅酶A脱氢酶β(hydroxyacyl coenzyme A dehydrogenase β, HADHB)。 人类 HADHB基因序列中含16个外显子。
冷冻电镜技术结果提示,人类HADHB与线粒体MTP的α-亚单位可组成α2β2四聚体,分子量约为460 kDa,这一结果对八聚体复合物的概念提出了质疑。复合物的中央为HADHB组成的二聚体,发挥支架作用,并与两侧α-亚单位相连接。两个α-亚单位在两侧呈反向存在,使得加水酶和脱氢酶对角相望。HADHB依赖α-亚单位连接于线粒体膜,且His379是酶活性的关键氨基酸残基,负责酯酰辅酶A的硫解反应[2]。
HADHB功能异常可造成人类线粒体脂肪酸β-氧化代谢障碍症。患儿出生后即可发病,主要病理与病理生理学变化包括心肌病伴随心律失常与心衰,肝肿大、肝功能障碍与肝脏脂肪变性,毛细血管通透性增大与水肿,后期神经系统与视网膜病变渐进性加重。患者血液生化指标可见13C-棕榈酸(C16酸)及13C-羟基化棕榈酸(13C-C16OH)高于正常,13C-乙酰肉碱的浓度则显著低于正常。亚裔群体存在点突变病例,错位位点有 c.739C>T,c.520C>T 及 c.1331G>A[3-4]。 β-氧化代谢障碍所造成的疾患不仅与细胞能量缺失有关,实验还观察到,长链3-羟脂肪酸的蓄积会降低线粒体膜电位,减弱线粒体的钙储存能力,促进线粒体肿胀,增加线粒体膜通透性转换孔的开放,进而造成细胞死亡[5]。
1.2 酶活性的调节 图1列出了参与HADHB调控的主要分子。采用免疫共沉淀及免疫荧光共定位等技术发现,雌激素受体-α与-β均可与HADHB相互作用,上调硫解酶活性,增加脂肪酸的β-氧化[6]。雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)不仅是细胞生长与增殖的重要因子,也参与脂肪酸氧化的调节。在其配体raptor敲除小鼠可见,心肌由脂肪酸代谢转向糖代谢,提供ATP;更为重要的是,诱导性敲除4周后心功能出现障碍。机制研究表明,HADHB等脂肪酸氧化及转运相关酶的表达明显减少;过氧化物酶体增殖物激活受体γ辅助因子-1α与-1β是线粒体氧化与能量生成的关键分子,实验表明,上述改变与这一分子无关[7]。
MicroRNA与HADHB的关系也有报道。实验发现,胆固醇调节元件结合蛋白(sterol-regulatory element binding proteins,SREBP)的内含子可转录miR-33a/b,miR-33a/b 直接抑制 HADHB 的表达[8]。 进一步研究表明,miR-33结合于3-UTR区,抑制基因表达[9]。 硫氧还蛋白结合蛋白(thioredoxin-interacting protein,TXNIP)可抑制该酶活性,其机制与miR-33也有关联。实验发现,敲除TXNIP可增加核因子Y(nuclear factor Y)与SREBP启动子的结合,减少miR-33a的转录,进而解除miR-33a对HADHB表达的抑制作用,上调HADHB的酶活性[10]。
MOXI(micropeptide regulator of β-oxidation)是一肌肉内富集的小分子多肽,由核基因编码,56个氨基酸组成。该分子定位于线粒体内膜,并与线粒体MTP相结合。基因敲除后可见,心肌线粒体以棕榈酸为底物的3态呼吸能力减弱,但以丙酮酸为底物的呼吸水平并未改变;离体灌注心脏表明,与野生型小鼠相比,组织利用脂肪酸能力减弱,利用碳水化合物的能力增加,而且小鼠运动后心肌储备能力明显减弱;补救实验可恢复脂肪酸的有氧氧化[11]。这些结果表明,MOXI是调节脂肪酸β-氧化的又一重要分子,其对HADHB的作用值得深入研究。
2 过氧化物酶体3-KAT
在过氧化物酶体,3-KAT并不与3-羟脂酰辅酶A加水酶及脱氢酶组成MTP,而是单独存在。按功能可将硫解酶分为两类,3-KAT负责降解长直链脂肪酸;另一类称为固醇载体蛋白-2(sterol carrier protein 2,SCP-2),其作用底物为支链脂肪酸。前者在啮齿类动物由A和B基因编码,分别命名为ThA与ThB。基因序列中均都含有12个外显子和11个插入基因,编码424个氨基酸。ThA主要表达于肝脏和小肠。ThB可见于肝脏,在肾,肠及白色脂肪组织中表达水平较弱。在人类,相应的基因目前仅找到一个,也被称为乙酰辅酶A酰基转移酶。SCP-2在人类和啮齿类动物均存在,因为基因序列中存在两个编码起始位点以及翻译后修饰,其产物有58 kDa、45 kDa及13 kDa三种形式,实验发现,前两者具有硫解酶活性。
目前对过氧化物酶体3-KAT的功能认识还知之甚少。Mandard课题组采用同源重组技术仅破坏酶的催化结构域,此功能缺陷小鼠未出现明显的表型异常。PPARα是促线粒体与过氧化物酶体氧化的关键分子,上调多种基因表达。给予ThB-/-小鼠PPARα激动剂Wy后,小鼠肝脏胆固醇浓度回升,涉及胆固醇合成的相关酶的表达明显增加,且这一作用不依赖于胆固醇调节元件结合蛋白[12]。该结果提示,3-KAT除了降解脂肪酸外,还参与胆固醇的生成,其分子机制有待阐明(图 1)。此外,Wy还提升了肝脏n-7、n-9单不饱和脂肪酸水平,表明3-KAT影响脂肪酸的组成(图1)。
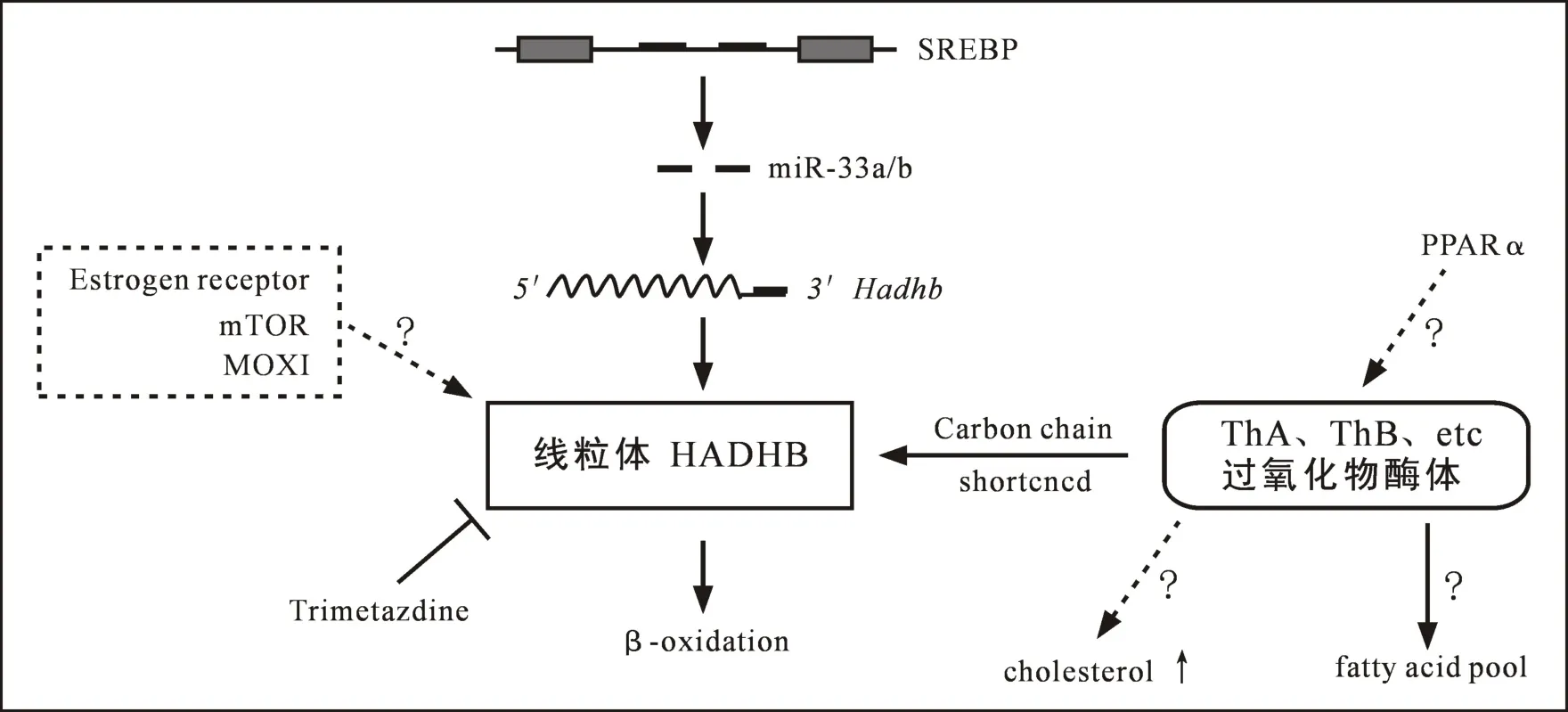
图1 3-酮脂酰酶辅酶A硫解酶的作用与调节机制模式图
3 3-KAT的药物靶向治疗
生理条件下,脂肪酸为心肌主要供能物质,提供的ATP约占总量的2/3。虽然在相同碳原子数量的情况下,脂肪酸产生ATP的数量高于葡萄糖;然而,脂肪酸的有氧氧化效率较低于葡萄糖,ATP/O的数值分别为2~2.5和3.17。 在心力衰竭患者,冠脉供氧不能满足心肌代谢需求,此时抑制长链3-KAT的活性,可以优化能量代谢,降低脂肪酸氧化,刺激葡萄糖氧化,将更有益于维持心肌功能[13]。曲美他嗪(trimetazidine,TMZ)是一种哌嗪类衍生物,具有良好的抗心衰作用。尽管其作用机制仍有争议,但学者Lopaschuk早在2003年即检测到,该药显著降低3-KAT对底物分子3-酮基-十六酰基辅酶A(3-KHCoA)的水解,说明曲美他嗪的抗心衰作用与其减弱脂肪酸代谢(图1),加强糖代谢密切相关。另有学者推测为,TMZ或许减弱骨骼肌脂肪酸氧化,进而增加胰岛素抵抗。然而实验结果恰巧相反,TMZ不但未增加,反而降低骨骼肌胞内游离脂肪酸浓度,提示整体情况下骨骼肌可能存在代偿作用,加速脂肪酸有氧氧化[14]。这使多数学者仍认为TMZ是一安全有效的改善心肌能量代谢的药物。
脂代谢障碍是动脉粥样硬化的病变基础,以miR-33为靶向的分子干预是一潜在的治疗新策略。细胞学研究发现,沉默miR-33将解除对HADHB等与脂肪酸氧化相关酶的抑制,同时增加胰岛素信号通路[9]。在低密度脂蛋白敲除小鼠观察到,沉默miR-33缩小粥样斑块,增加细胞内胆固醇外排[15];后续在高脂饮食小鼠检测到,沉默miR-33将上调Hadhb的表达,改善线粒体功能,加速脂肪酸氧化[16]。这些结果提示,上调Hadhb或为沉默miR-33抗动脉粥样硬化的重要下游事件之一。同时,上调miR-33,抑制Hadhb,进而减弱脂肪酸氧化可否成为慢性缺血心肌保护策略有待研究。
小结:3-KAT是脂肪酸β-氧化的关键分子,实验发现,多种因子参与对其活性的调节,提示机体可通过不同的方式调节脂肪酸氧化以适应代谢需要,然而其作用机制尚不清楚。此外,人类过氧化物酶体中该酶的作用尚不清楚。因此,深入阐明3-KAT的功能,相应的信号通路及作用机制对临床相关疾病的防治有着重要的意义。
猜你喜欢
黑龙江大学自然科学学报(2022年4期)2022-11-17 08:07:52
河北科技师范学院学报(2022年2期)2022-08-26 08:55:46
中老年保健(2022年4期)2022-08-22 02:58:30
海洋通报(2021年1期)2021-07-23 01:55:14
生物学通报(2021年4期)2021-03-16 05:41:26
中国塑料(2016年3期)2016-06-15 20:30:00
中外医疗(2015年11期)2016-01-04 03:58:49
华东理工大学学报(自然科学版)(2015年4期)2015-12-01 04:00:36
癌变·畸变·突变(2014年1期)2014-03-01 04:39:36
中国中医药现代远程教育(2014年23期)2014-03-01 04:33:21
