
JIVE方法在卵巢癌多组学数据整合分析中的应用*
2019-11-12 12:24:24哈尔滨医科大学卫生统计学教研室150081
中国卫生统计 2019年5期
哈尔滨医科大学卫生统计学教研室(150081)
徐 欢 宋 微 蔡雨晴 侯 艳 李 康△
【提 要】 目的 引入JIVE方法对多组学数据进行整合分析,并应用于肿瘤分子分型研究。方法 使用TCGA数据库中卵巢癌mRNA和miRNA的组学数据,应用JIVE方法整合分析两个组学数据,提取两不同组学数据的共同特征,然后通过对其具有共同结构的数据做主成分分析,给出卵巢癌miRNA分子分型的结果。结果 经过JIVE方法整合分析后,使miRNA数据具有明显与mRNA相一致的分型结构,从而进一步支持了mRNA的分型结果,同时揭示了两组学之间在组织分子分型上具有一定的调控关系。结论 JIVE方法可以用于提取组学之间存在的共同结构矩阵,从而进行多组学数据的整合分析。
组学数据(omics data)是指通过测序仪、生物芯片、磁共振、色谱-质谱联用等高通量实验技术得到的基因组、转录组、蛋白质组和代谢组等数据。通过对高维多组学进行整合分析,可以研究相关疾病的分子分型及标志物,对疾病的早期诊断、临床治疗和预后具有重要意义。本文引进JIVE(joint and individual variation explained)方法[1],将其应用于卵巢癌mRNA和miRNA两组学数据的分析,揭示两种组学之间潜在的生物学关系,为研究其分子分型的调控机制提供有价值的分析结果。
原理和方法
设有k个组学数据矩阵X1,X2,…,Xk,分别为p1、…、pk行和n列,其中行为各组学数据的变量,列为样本。JIVE方法的基础是矩阵的奇异值分解,即将不同组学的数据矩阵合并后分解为三部分,即描述不同组学数据的共同结构矩阵(J)、独立结构矩阵(A)和残差矩阵(R)。具体步骤如下:
1.数据预处理
首先将多个矩阵按行合并,合并的数据矩阵:
(1)
为了使每个数据集对合并矩阵的总变异贡献相等,需要对其标准化,首先对其中心化去除不同数据集的基线差异。
(2)
然后再将数据矩阵归一化,使合并矩阵中每个不同组学的数据集的总变异贡献相等,对此可以应用Frobenius范数:
(3)
归一化的数据矩阵为
(4)
(5)
对合并矩阵可以做如下奇异值分解
2.确定共同矩阵和独立矩阵的秩


(6)
通过最小化残差平方和来确定共同结构与独立结构,即给定矩阵的秩,使‖R‖2最小来确定J和A1,…,Ak。这一步通过一个迭代运算来完成,对于第t次迭代有
(7)
其中R是一个p×n的残差矩阵,重复以上步骤直到找到最合适的J和A1,…,Ak使‖R‖2最小。

实例分析
研究背景:2011年TCGA团队对TCGA中489例卵巢癌mRNA数据进行聚类分析,结合专业得到了四个分型,即增殖型、间叶细胞型、分化型和免疫反应型,该结果发表在Nature杂志上[3]。本例主要分析miRNA是否能够做同样的分型及是否能够说明其在分型上与mRNA具有潜在的调控作用。
使用TCGA卵巢癌基因表达数据mRNA与miRNA数据(经标准化),进行样本ID匹配后,得到样本量为408的样本。其中mRNA基因数目为20113个,miRNA的数目为680个,共两个数据矩阵。首先对数据进行预处理,去除标准差较小的mRNA,对mRNA设定的阈值为SD≤1.5;与mRNA相比,miRNA具有更强的时空表达异质性,即在一些位置和时间不表达,因此剔除零表达个数超过样本量一半的miRNA。预处理后得到mRNA表达矩阵为302行,408列,miR-NA表达矩阵为351行,408列。将两个数据矩阵合并后进行JIVE方法分析,得到共同结构矩阵秩为3,mRNA独立结构矩阵秩为34,miRNA独立结构矩阵秩为23。共同结构的解释方差及置信区间分别为1.64×10-9(1.48×10-9,1.78×10-9)、2.35×10-9(2.16×10-9,2.52×10-9),独立结构的解释方差及置信区间分别为4.62×10-9(4.43×10-9,4.78×10-9)、3.18×10-9(3.01×10-9,3.35×10-9)。JIVE分解可以得到的三个矩阵,解释方差占比如图1。

图1 JIVE分解得到三部分解释方差比
分别对数据矩阵X2和共同结构矩阵J2做主成分分析,样本前三个主成分得分图如图2。结果显示,对于miRNA数据,原始矩阵X2主成分解释方差占比分别为10.30%、8.62%、6.86%,散点图中各点混在一起,而其共同结构矩阵J2则呈现出与mRNA表达分类相同的分类趋势(主成分解释方差占比分别为55.93%、27.11%、16.96%),说明使用JIVE分解方法可以看到两组潜在的调控关系。

图2 mRNA及JIVE分解前后miRNA表达数据PCA分类图对比
为了显示其综合分类效果,将JIVE分解得到的共同结构矩阵J1和J2合并在一起做主成分分析,使用前两个主成分分别给出其样本得分的密度分布图,参见图3和图4,变量的因子载荷见表1(mRNA、miRNA按照绝对值排序各前5位)。
结果显示,第一主成分的样本得分可以区分增殖型与间质型样本,第二主成分的样本得分可将增殖型、间质型与另外两种分子分型区分开。计算各共同成分的变量载荷,第一主成分中因子载荷绝对值由大到小前五位的基因分别为SFRP2、POSTN、DLK1、LUM、MMP11,其中基因SFRP2的甲基化与卵巢癌患者复发和生存率相关,POSTN基因高表达的卵巢癌患者总生存期和一线化疗后的无进展生存期明显更短,DLK1在浆液性卵巢癌和浆液性交界性癌中均高表达,第一主成分中因子载荷绝对值由大到小前五位的miRNA分别为miR-508、miR-202、miR-509,其中miR-508、miR-509的高表达与卵巢癌患者更长的生存期有关,第二主成分因子载荷较大的变量中,IGF2的高表达与晚期卵巢癌的发生发展和化疗药物耐药性有关,CLDN6在卵巢癌组织中表达上调,COL3A1是能够独立预测卵巢癌铂基化疗耐药性的基因,miR-483-3p、miR-370与卵巢癌的耐药机制及化疗敏感性相关。
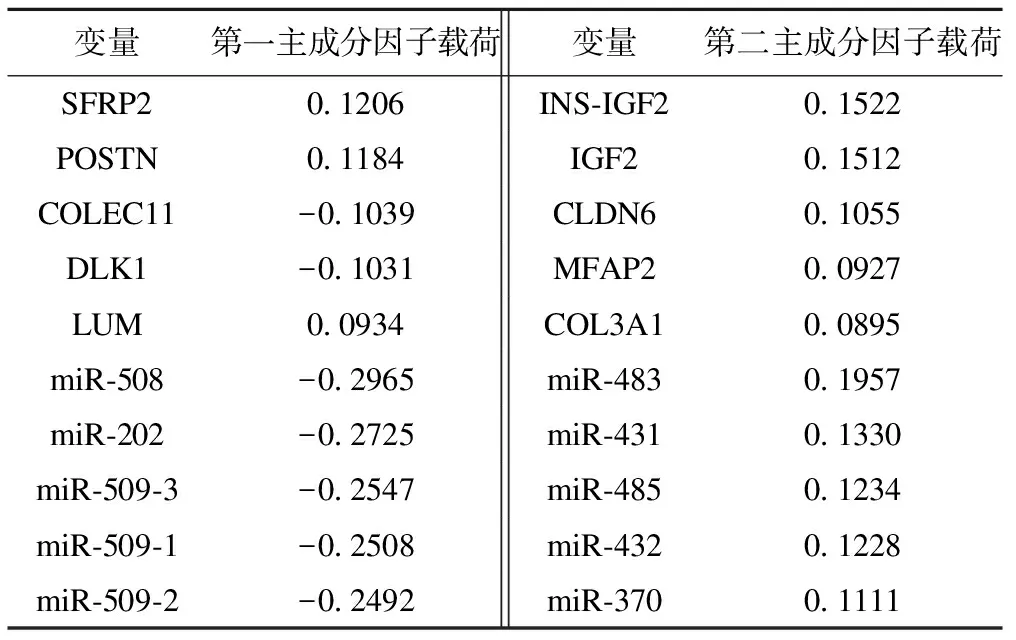
表1 变量在前两个主成分中的因子载荷
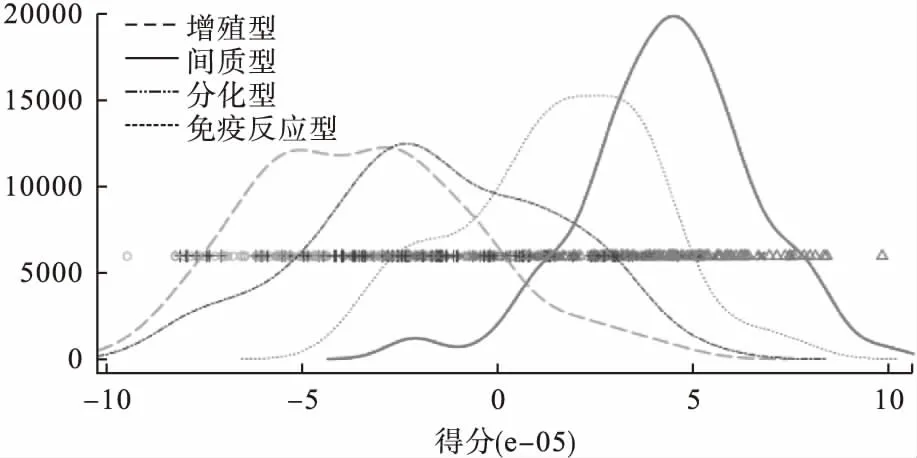
图3 第一主成分的样本得分概率密度分布图
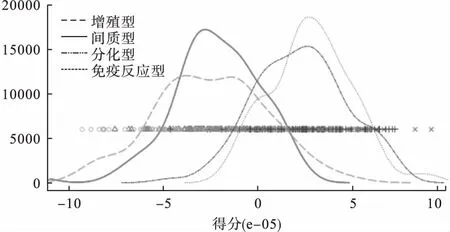
图4 第二主成分的样本得分概率密度分布图
讨 论
分子分型在临床实际中具有重要意义,如不同分型对特定的药物敏感性不同或预后不同[4]。本研究根据由mRNA得到的4个卵巢癌分子分型引入了JIVE方法分析卵巢癌miRNA数据,提取miRNA与mRNA的共同结构,得到了共同结构的分类特征,揭示了miRNA在分子分型上是否与mRNA存在可能的调控关系。
JIVE方法使用的前提是不同组学数据间有足够的共性可以提取。一般来说,共同结构矩阵的秩决定了不同组学之间调控关系的复杂程度,秩越大,表明数据可能具有更复杂的潜在分型结构。而对于提取的共性结构如何解释则需要结合生物学背景知识。如,本文实例中提取的共性结构表示卵巢癌miRNA的表达与分子分型之间存在相关关系。需要注意,计算秩的方法是置换检验,因此当样本量过小时,亦不容易得到显著的秩,亦难以获得组学之间的关联信息,其他计算秩的方法仍需进一步研究。另外,JIVE对缺失数据不稳健,应先选择合适的方法进行填补再分析。
奇异值分解的原理与PCA相似,只有在数据中的方差贡献足够大时,其特征才能够被提取出来,因此需要舍弃那些方差贡献小的变量,尽量减少对分析的干扰,以获得更好的分析结果。本研究使用的数据粗筛的方法是“去除SD<1.5的mRNA变量和“0”表达个数超过总样本量的一半的miRNA变量”,实际中也可以使用其他的阈值或方法。用敏感度分析的思路去考察粗筛方法对结果的影响,结果差别不大。
猜你喜欢
基层中医药(2020年5期)2020-09-11 06:32:00
家庭医学(下半月)(2020年2期)2020-05-11 02:07:26
国际口腔医学杂志(2019年3期)2019-05-31 10:09:26
基层中医药(2018年5期)2018-08-31 02:35:42
天然产物研究与开发(2018年2期)2018-04-04 02:01:12
安徽医科大学学报(2016年12期)2017-01-15 14:21:54
医学研究杂志(2015年11期)2015-06-10 06:44:03
医学研究杂志(2015年4期)2015-06-10 06:42:43
制造技术与机床(2015年10期)2015-04-09 07:06:14
中国中医药现代远程教育(2014年21期)2014-03-01 04:32:23
