
单细胞RNA测序在肾脏疾病研究中的应用
2019-10-22 09:16陈麒麟综述刘志红审校
肾脏病与透析肾移植杂志 2019年4期
陈麒麟 综述 刘志红 审校
DNA序列、染色质结构、RNA转录水平、蛋白质和代谢物等大规模的组学研究,为生命与疾病过程进行了丰富与全面的描述[1]。然而传统转录组学研究(Bulk RNA-seq)只能应用于由许多细胞组成的组织样本,提供均值化宏观数据的同时掩盖单个细胞间的差异,缺乏针对单个细胞的认识。单细胞RNA测序(scRNA-seq)是一项获取单个细胞转录水平的研究方法,能够从单个细胞层面进行组织或器官的研究。随着单细胞技术的发展,2016年10月科学界正式发起了“人类细胞图谱”(human cell atlas,HCA)的国际合作研究计划,其基本目标是采用特定的分子表达谱来确定构成人体的所有细胞类型[2]。大规模scRNA-seq实验目前已获得了线虫、蜗虫、果蝇和小鼠大多数器官的细胞图谱。在人类研究中,单细胞分析提高了对发育、老化和癌症等不同病理改变的认识[3]。
肾脏细胞的形态与功能存在较大异质性。通过显微微分离等方法已实现了肾小球与肾小管的区域性组学研究,证明了肾小球与肾小管细胞上的显著差异[4]。但同一区域的肾脏细胞同样存在明显差异,如肾小球中终末分化状态的上皮细胞(足细胞)、维持血管基本结构的内皮细胞和血管周围的系膜细胞或者组成肾小管各个节段的各种上皮细胞,都拥有独特的形态特征,发挥各自特异的功能,在不同疾病中存在不同的损伤改变[5]。
scRNA-seq应用于肾脏领域能够极大地提高对于肾脏细胞异质性的认识,本文就scRNA-seq技术的基本原理及其在肾脏疾病的应用进行综述。
scRNA-seq的基本技术
转录组测序是分析基因表达的有效方法,主要过程包括将RNA逆转录为cDNA,然后进行高通量DNA测序,序列读取的数量能反映样本中基因的表达水平。scRNA-seq的基本原理与之类似,不同之处在于其针对单个细胞进行研究,使其在单细胞悬液制备、单细胞捕获技术、数据处理与分析中具有一定特殊性。
单细胞悬液制备单细胞悬液中细胞状态及细胞内RNA的完整性直接决定单个细胞的有效捕获与研究结果的真实性。血液等天然单细胞悬液可经过梯度离心等方法直接进行单细胞悬液制备,而肾脏等实体组织必须通过机械分离和消化酶处理。目前认为应根据不同组织采用不同消化酶进行不同时长的处理[6]。肾脏组织的复杂性使其在单细胞悬液制备方面存在一定难度,尚缺乏公认的标准。
此外,样本处理可能影响细胞状态及基因表达。研究证实消化酶会激活应激相关基因[7]。研究提示低温酶消(4℃)可有效降低常规37℃生物酶消对于细胞的影响[8]。研究发现破坏细胞膜获取单个细胞核进行单细胞核转录组测序(snRNA-seq)在肾脏scRNA-seq中具有一定优越性[9]。但细胞RNA中仅10%~20%位于细胞核内,snRNA-seq将丢失细胞质RNA信息[10]。
单细胞捕获技术捕获单个细胞并对单个细胞的RNA进行扩增是scRNA-seq的特色。单细胞捕获方法可以分为孔板与微流控技术,RNA扩增方法可分为指数扩增与线性扩增。目前scRNA-seq根据单细胞捕获的方法不同,细胞RNA扩增方法的不同分为不同方法。下面就几种经典的scRNA-seq方法进行介绍。
通过人工吸取、激光显微切割和流式细胞仪分选等方法,研究者可将单个细胞捕获至孔板中,裂解孔板中的单个细胞,在独立封闭的反应孔中进行后续扩增。
Smart-seq2是一种通过模板置换扩增单细胞全长转录本的方法,能够获取单个细胞1万多个基因的表达,此方法被广泛改良并运用于多种scRNA-seq方法中[11]。STRT-seq在Smart-seq2的基础上引入一段特定序列(单细胞条形码 barcode)合并到细胞cDNA序列中,同一细胞的cDNA通过复制扩增出具有相同barcode的序列,实现将多个细胞的cDNA序列进行合并处理。STRT-seq丰富了5′端转录本,使其具有提供转录起始位点信息的优势[12]。CEL-seq使用包含T7启动子的poly (T)引物实现cDNA的线性扩增,产生更均匀的扩增结果[13]。MARS-seq在poly(T)引物中加入特异分子标志(UMIs)并通过自动化流程提高了细胞数量[14]。纳米级微孔板将试剂反应体积从微升降低到纳升,降低成本和实验不稳定性。基于纳米孔的Seq-Well可捕获86 000个细胞[15],单细胞悬液中的单个细胞因重力作用自然沉降到纳米微孔,在独立微孔中进行后续标记与扩增。郭国骥团队研发的Microwell-Seq采用类似的技术原理,采用琼脂糖制备微孔板,保证稳定性的同时,降低实验成本。该团队通过此方法首次绘制了小鼠大部分器官与组织的细胞图谱[16]。
以集成流体管道(IFCs)和微液滴为代表的微流控系统是另一种单细胞捕获技术。Fluidigm C1是第一个用于scRNA -seq的微流控系统。单个细胞随着液体进入不同孔径的芯片,固定在芯片的纳米级反应空间内,通过改良的Smart-seq方法进行扩增建库。改进后的实验方案将细胞量从96个增至800个(C1 HT-IFC),实现细胞的混合处理[17]。另一类微流控技术通过产生大量纳米级微液滴,形成油包水结构,将细胞封闭在液滴中,使其具有独立的反应空间,技术方式包括Drop-seq[18]和InDrops[19]。Drop-seq中细胞RNA与附着于磁珠的barcode结合,在一个共同的反应管内进行逆转录。而对于InDrop,barcode与mRNA杂交后直接在液滴中进行逆转录反应。研究显示InDrops能对50%的上样细胞产生测序数据,其成功率是Drop-seq的十倍[10]。10x Genomics公司根据InDrops进行一定修改后开发出Chromium单细胞测序平台,该平台目前已广泛应用于各类单细胞研究中[20]。
数据处理与分析scRNA-seq数据处理与分析的方法与Bulk RNA-seq类似。不同的是单细胞数据涉及将条形码、UMI数据与转录本数据拼接对齐,单个细胞转录组相似性聚类分析等问题。
scRNA-seq数据在进行分析之前需要良好的质量控制,应注意排除红细胞污染、双细胞、细胞碎片干扰等数据。细胞线粒体RNA含量较高可能表示该细胞结构已出现严重破坏,细胞丢失大量细胞质RNA,不能反映细胞真实情况[10]。但应注意甄别部分能量代谢旺盛的细胞中线粒体RNA比例较高的情况[21]。
scRNA-seq数据分析得到多个细胞的基因表达谱,可通过细胞多个基因的表达水平进行细胞聚类。细胞多基因相似性代表细胞多维度的相对关系。研究者通常采用降维处理的方法,将多维关系较为真实地投射到二维或三维体系中,细胞在二维图谱中的相互距离表示其在基因水平的差异程度。主成分分析(PCA)和t分布随机邻域嵌入分析(t-SNE)是常用的降维方法[22]。注释聚类后驱动集群的基因(标记基因),或对该亚群细胞进行功能分析,可反映细胞亚群的生物意义。此外通过伪时间分化分析的方法可模拟多个亚群细胞的动态分化或转变过程[23]。
机器学习等人工智能领域的快速发展,使得单细胞测序的数据分析方法不断丰富,能够更加全面、准确地阐释scRNA-seq数据。
正常肾脏细胞图谱
随着scRNA-seq的迅猛发展,研究者逐渐认识到scRNA-seq对于肾脏研究的重要性。本中心首次针对20个正常小鼠足细胞[24]和33个系膜细胞进行scRNA-seq研究[25]。这是首次对肾脏固有细胞基因表达情况的单细胞研究。宾夕法尼亚大学团队对7只健康雄性小鼠肾脏进行scRNA-seq研究,将43 745个细胞进行聚类分析,确定了16个不同的细胞亚群,通过伪时间分化轨迹分析和体内谱系追踪发现了一种处于闰细胞和主细胞之间的新的细胞亚群。研究还证明了Notch通路的激活可驱动闰细胞向主细胞的转变。这项工作首次描绘了正常成年小鼠肾脏细胞的完整基因表达图谱,并且展现出成熟肾脏惊人的分化灵活性[21]。另一团队通过癌旁正常肾组织捕获了37 951个正常人肾脏非免疫细胞和4 796个正常肾脏免疫细胞,绘制了人的正常肾脏细胞图谱[26]。肾脏scRNA-seq研究使人们对于肾脏细胞的认识与分类从形态学逐渐发展到分子水平。
目前在正常肾脏scRNA-seq数据中,以近端肾小管上皮细胞为主[27]。之前根据肾脏不同解剖分区的研究已经证明了区域研究能够有助于对肾小球的认识[4]。研究者针对正常小鼠肾小球进行scRNA-seq,在每个细胞平均630个基因的测序深度上,得到近13 000个单细胞转录组数据,鉴定出五种肾小球亚群:足细胞(80%)、系膜细胞(2%)和内皮细胞(12%)、小管细胞(6%)和免疫细胞(0.2%)[28]。尽管这样的细胞种类构成可能受到组织消化等因素影响,但研究仍发现内皮细胞中有一群增殖相关的亚群,足细胞亚群更为均匀,可大致分为三个以前未知的亚群。
由于缺乏跨物种相似细胞群的共同标记,使得从动物模型向人类肾脏研究的转化较为困难。阿拉巴马大学团队通过分选小鼠、大鼠、猪和人的肾脏免疫细胞后进行scRNA-seq,发现CD74和CD81为肾脏巨噬细胞跨物种的标志物[29]。该研究丰富了针对肾脏定居免疫细胞的研究方式,也为多物种肾脏研究及临床转化提供了新的研究思路。
肾脏疾病的scRNA-seq研究
通过scRNA-seq研究,人们希望了解肾脏细胞在疾病过程中的病理改变过程,更加精细地展现肾脏疾病的发病机制,并为精准靶向逆转和改善疾病发展过程提供可能的方向(图1)。
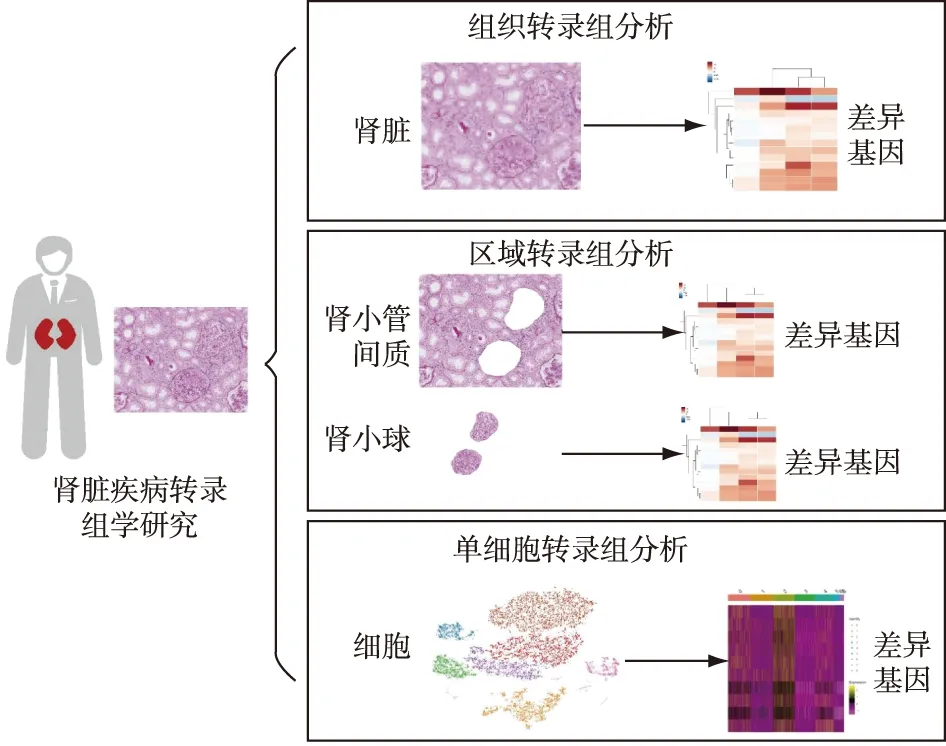
图1 肾脏疾病单细胞RNA测序研究示意图
本中心合作开展了针对糖尿病肾损害小鼠(DKD)肾小球的scRNA-seq研究,DKD肾小球可分为五种细胞群,包括肾小球内皮细胞、系膜细胞、足细胞、免疫细胞和部分肾小管细胞。研究发现DKD肾小球的免疫细胞数量明显多于正常肾小球,而这些免疫细胞以巨噬细胞为主。通过与对照组小鼠内皮细胞和系膜细胞比较发现,DKD的内皮细胞中血管生成和迁移通路的基因和系膜细胞中调控基因翻译、表达及蛋白稳定的基因均发生改变,并在Bulk RNA-seq中得到证实[30]。
Der等[31]对狼疮性肾炎(LN)患者的皮肤、肾活检样本及外周血进行了scRNA-seq研究。该研究发现一种以干扰素反应相关的基因与LN严重程度有关。LN患者的皮肤样本表现出该基因的类似特征,表明这种更易获取的组织样本一定程度上可以反映LN患者的疾病状态。
从小鼠肾脏scRNA-seq的数据发现,人类单基因遗传疾病中表现为蛋白尿的有21个基因在小鼠中只表达于一类细胞——肾小球足细胞[21]。尽管有研究认为内皮细胞和近端小管在蛋白尿的形成过程中存在一定关系,但本研究表明足细胞功能障碍是蛋白尿发生的主要原因。该研究还发现导致肾小管酸中毒的基因只表达于小鼠集合管闰细胞,证明了该细胞在酸碱平衡中的重要作用。进一步通过对血压、慢性肾脏病、血清代谢物水平和肾结石相关的疾病基因进行分析,发现大多数疾病相关基因只在单个细胞类型中表达。该研究通过scRNA-seq发现导致肾脏疾病的基因主要位于一种特定类型的细胞,这提示肾脏某一细胞种类的病理改变可直接导致特定疾病的发生[21]。
针对以混合排斥反应为主的移植肾scRNA-seq研究发现,移植肾单核细胞的浸润在排斥反应中发挥关键作用。与正常对照相比,有两群单核细胞在正常肾脏中表达较少,而在移植肾混合排斥反应表达增加。一群表达FCGR3A (CD16)的单核细胞与CD16阳性、促炎、非典型单核细胞相似,另一群则类似经典或中间态单核细胞。该研究进一步对成人肾脏进行snRNA-seq,在4 259个单细胞核数据中确定了6种不同的上皮细胞亚群,包括足细胞、近端小管、主细胞和闰细胞等。通过整合scRNA-seq与snRNA-seq数据,将排斥反应与正常对照组织对比,发现排斥反应并未从根本上改变肾小管细胞的特性。此外还发现内皮细胞分为三个不同的亚群:静止细胞亚群和两种活化的内皮细胞群。其中,一种活化的内皮细胞亚群表达Fc受体通路激活的基因,与抗体介导排斥反应的病理诊断结果一致[32]。
scRNA-seq研究还涉及肾脏肿瘤中异质性明显的肿瘤细胞和浸润肿瘤的免疫细胞。对于转移性透明细胞癌的研究发现,转移的肿瘤细胞对靶向表皮生长因子受体(gefitinib、erlotinib和afatinib)、SRC家族激酶(dasatinib)和BRAF-MEK (selumetinib)的药物敏感,而原发肿瘤细胞对靶向MET的药物(tivantinib、foretinib和crizotinib)和磷酸肌醇激酶(BKM120)更敏感,证明利用scRNA- seq研究可以为患者提供更加合理的个体化诊疗方案[33]。一项来自人类肾脏肿瘤、正常胚胎,儿童和成人肾脏的scRNA-seq研究,证明Wilms肿瘤细胞来源于异常胚胎细胞,揭示了人类肾脏肿瘤的精确细胞特征和组成,为未来肾脏肿瘤研究提供了方向[26]。一项对73例肾透明细胞癌患者和5例健康对照者的研究,在350万个细胞中确定了17个肿瘤相关巨噬细胞亚群,22个T细胞亚群,以及一个与肿瘤无进展相关的独特免疫细胞组成,展现了肾脏肿瘤微环境的免疫细胞图谱,揭示了免疫治疗的潜在生物标志物和靶点[34]。
scRNA-seq在肾脏应用的挑战与展望
肾脏细胞中上皮细胞众多,细胞外基质结构复杂,如何从肾脏复杂环境中获取状态良好的单个细胞是目前肾脏scRNA-seq研究所面临的一个难题。普遍认为单个细胞表达的基因数应在5 000~20 000,而目前大部分高通量scRNA-seq方法很难实现如此高的基因捕获率,致使部分转录信息被遗漏。低覆盖的单细胞转录组数据可能直接导致错误的细胞聚类,从根本上削弱scRNA-seq的研究意义。虽然存在单细胞制备困难、微量RNA扩增偏倚等情况,但是迅猛发展的scRNA-seq研究已经在肝脏、脑、脾、肺、胎盘等组织器官中得到了广泛应用,并取得了令人瞩目的结果。
随着scRNA-seq研究的发展与革新,单细胞基因组学、单细胞表观遗传组学、单细胞蛋白组学及单细胞空间组学也逐渐发展与丰富[20]。其中单细胞空间组学能够对切片组织进行原位RNA水平或蛋白质水平的定量分析,这可能极大改变目前对于肾活检组织的评价方式[35]。通过整合单细胞组学技术,我们可以在单个细胞水平对疾病过程进行全面了解。我们有理由相信scRNA-seq所带动发展的单细胞多组学研究能够极大提升目前对于肾脏的认识,推动实现肾脏领域精准医疗的进程。
猜你喜欢
中华骨与关节外科杂志(2022年1期)2022-08-31
中国医药科学(2022年5期)2022-05-05
世界科学技术-中医药现代化(2021年8期)2021-12-21
天津医科大学学报(2021年4期)2021-08-21
科学(2020年4期)2020-11-26
教育界·上旬(2020年8期)2020-06-27
江苏农业科学(2017年16期)2017-10-27
中国卫生标准管理(2015年3期)2016-01-14
医学研究杂志(2015年9期)2015-07-01
中国药业(2014年24期)2014-05-26
