
自体动静脉内瘘功能障碍的机制
2019-10-22 09:16董建华综述葛永纯审校
肾脏病与透析肾移植杂志 2019年4期
董建华 综述 葛永纯 审校
血液透析(HD)是终末期肾病(ESRD)患者主要的肾脏替代治疗方式,功能良好而持久的血管通路是患者顺利接受HD的必备条件,因此,血管通路是HD患者赖以生存的生命通道。NKF/KDOQI指南推荐自体血管动静脉内瘘(AVF)作为HD患者首选血管通路,其次为移植物血管,最后选择为中心静脉导管。血管通路功能直接影响HD患者的生存质量,通路功能障碍一直是困扰肾脏科医师的重要临床问题。
AVF功能障碍由一系列进行性的血管损伤因素所致,主要包括[1]:(1)尿毒症毒素损伤血管;(2)AVF建立后血流动力学变化;(3)内皮细胞功能障碍、炎症反应、氧化应激等因素引起血管狭窄和血栓形成;(4)经皮腔内血管成形术(PTA)时球囊扩张机械性损伤导致血管再狭窄(图1)。
血管狭窄和血栓形成是导致所有类型血管通路功能障碍的最常见原因,然而,由于目前对内瘘功能障碍的病理生理机制尚缺乏足够认识以及有效干预措施,因而,本文对自体动静脉内瘘血管狭窄的组织病理学特征、血流动力学因素和分子生物学机制在血管通路功能障碍中的作用进行综述,以进一步提高对自体动静脉内瘘功能障碍的认识。
ESRD患者血管的病理改变
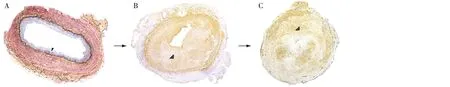
图1 血管通路功能障碍进程
ESRD患者大多存在高血压病、糖尿病、系统性红斑狼疮、ANCA相关血管炎等基础疾病,加之尿毒症毒素所致炎症和氧化应激损伤,在建立血管通路时,这些患者的血管组织往往已存在病理性改变,即内膜增生、中膜纤维化和微钙化。一项AVF队列研究在手术中留取554例静脉组织标本,病理结果显示88%的尿毒症患者存在静脉内膜增生,多为不规则增厚,其中57%患者内膜增生程度超过管腔面积的20%;免疫组化染色提示增生内膜主要由细胞成分和细胞外基质构成,其中细胞成分以肌成纤维细胞和平滑肌细胞为主,伴少量单核巨噬细胞浸润,细胞外基质主要由蛋白聚糖和少量胶原蛋白构成;此外,82%静脉中膜中存在大量胶原蛋白成分[2](图1A)。尽管如此,大多研究显示术前静脉内膜增生与AVF成熟障碍无关[4-5];当动脉直径≥2 mm时,动脉内膜增生与AVF术后6周内内瘘血流量、直径和内瘘成熟障碍亦无相关性[6]。
血管中膜纤维化主要表现为Ⅰ型和Ⅲ型胶原蛋白沉积,血管硬度增加,其与血管扩张和AVF成熟相关性尚存在争议。Martinez等[5]发现术前静脉中膜纤维化与AVF成熟无关;Allon等[7]亦未观察到术前动脉中膜纤维化限制AVF成熟。然而,Shiu等[8]研究发现AVF术前动脉中膜纤维化增加内瘘直径和血流量,促进内瘘成熟,可能与中膜纤维化增加内瘘压力有关。
血管钙化主要累及内膜和中膜,动脉粥样硬化以内膜钙化为主,而糖尿病和慢性肾脏病(CKD)患者中膜钙化更为突出。AVF术前桡动脉和肱动脉中膜微钙化普遍存在[9],而静脉全层均可出现不同程度的血管微钙化,主要位于中膜和内膜[10]。Allon等[11]发现AVF术前动脉微钙化不能预测AVF成熟障碍,但Choi等[12]观察却发现动脉微钙化与AVF术后1年内内瘘失功有关。因而,术前血管内膜增生、中膜纤维化和微钙化对AVF的影响尚存在分歧,仍需进一步明确。
血管通路狭窄的组织病理学特征
AVF成熟过程中,内瘘静脉会发生血管壁增厚和管腔扩张两个重要变化。内瘘狭窄和血栓形成是AVF功能障碍的最主要原因,而动静脉吻合口及吻合口旁静脉则是最常见的狭窄部位。
静脉内膜异常增生和血管外向扩张重塑障碍是引起内瘘管腔狭窄的主要病理改变。静脉狭窄区域的组织病理学表现为偏心性新生内膜增生,细胞成分以肌成纤维细胞为主,收缩性平滑肌细胞较少[13](图1B)。血管向外扩张重塑在AVF成熟中至关重要,静脉中膜纤维化与血管僵硬有关,影响血管重塑。Martinez等[5]通过一期AVF手术(获取术前静脉)和二期转位手术(获取内瘘静脉),观察静脉中膜纤维化和内膜增生与AVF成熟的关系。术后静脉中膜纤维化程度与AVF成熟不良相关,内膜增生则与之无关;而中膜纤维化严重的静脉(术后中膜纤维化程度≥48%),内膜增生可导致AVF狭窄。静脉向外扩张重塑可能与静脉中膜胶原纤维排列有关。Martinez等[5]将纤维排列整齐程度定义为各向异性指数(0-完全随机,1-排列整齐),各向异性指数小提示纤维排列紊乱,年龄大者各向异性指数小,AVF术前各向异性指数0.21±0.08与术后0.21±0.09相近。将胶原纤维走向与血管轴夹角定义为纤维定向角(0°平行,90°垂直),纤维定向角大者易导致静脉向内重塑(静脉舒张不足),女性和有AVF手术史者的纤维定向角大;术后纤维定向角和AVF成熟不良相关(图2)。
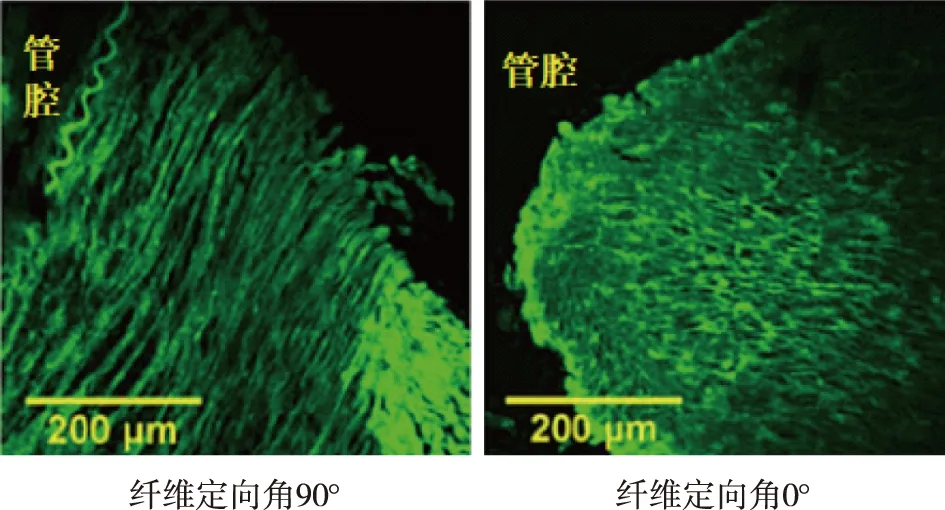
图2 自体动静脉内瘘静脉中膜胶原纤维排列(二次谐波发生成像)[9]
PTA是血管通路狭窄的主要治疗方法。PTA通过球囊扩张,使静脉内膜-中膜破裂,解除血管通路狭窄,引起血管重塑。PTA损伤血管内皮细胞和平滑肌细胞,引起内皮细胞和平滑肌细胞增殖,以及新生内膜增生,再狭窄处较初次狭窄内膜增生更明显[3](图1C)。
血管通路的血流动力学变化
正常静息状态下肱动脉平均血流量约50 ml/min,桡动脉通常小于25 ml/min,头静脉血流量几乎不可测得。人体正常血管内血流多为层流,血管通路建立破坏了供血动脉压力梯度的连续性,在局部创造了一个高顺应性、低阻力的血流动力学环境,血流表现为湍流。成熟内瘘血流量大于500 ml/min,血管因管腔内高血流量、高压力变化产生结构性适应性改变称为血管重塑。血管壁剪切力和环向力是影响血管重塑的重要机械性因素。
血管壁剪切力是血液流动对血管壁的摩擦力(单位dyne/cm2)。人体动脉生理状态下剪切力在10~70 dyne/cm2。生理范围内的剪切力(15~50 dyne/cm2),使血管内皮细胞相对静止,内皮细胞顺血流方向生长和排列,分泌抗炎和抗凝血物质,内膜增生受抑制,管腔内径增加,血管良性重塑。非生理性剪切力,即低/振荡剪切力和高剪切力,均会影响血管重塑。剪切力<10 dyne/cm2或振荡剪切力的湍流,可导致内皮细胞活化、增殖,细胞形态紊乱,分泌炎症和凝血物质,引起内膜异常增生,血管外向重塑障碍,血管内径狭窄,血栓形成,导致AVF功能障碍[14](图3)。而剪切力>70 dyne/cm2时,则会刺激内皮细胞增殖并诱导血栓形成[15]。
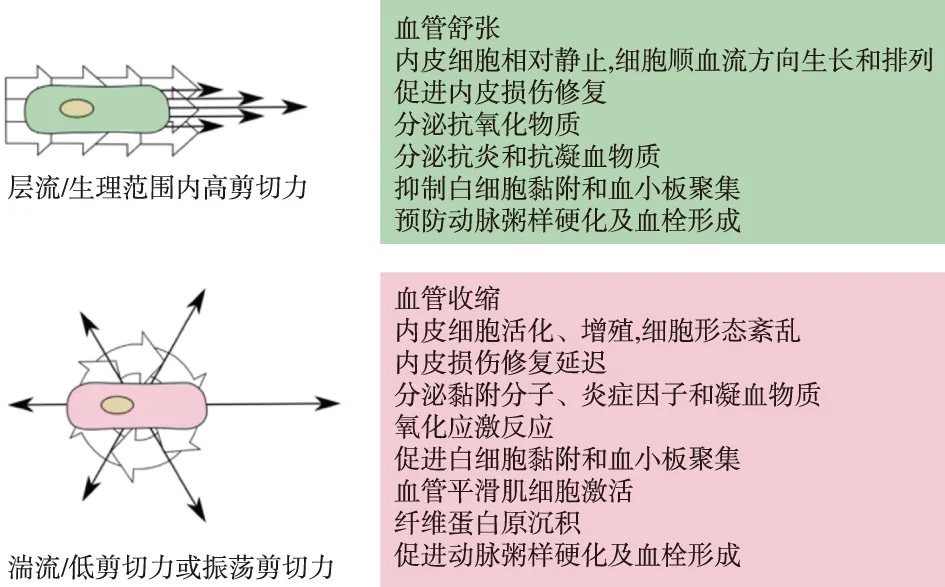
图3 不同血流动力学及剪切力对血管内皮细胞的影响
Corpataux等[16]发现6例患者AVF术前头静脉剪切力为5~10 dyne/cm2,术后最高可升至24.5 dyne/cm2,血管重塑过程中,内瘘静脉直径逐渐增大,剪切力在术后12周逐渐下降至10.5 dyne/cm2。Rajabi-Jagahrgh等[17]研究进一步证明,无论AVF剪切力初始值多少,均会随时间而下降;剪切力下降与静脉外向重塑和AVF成熟相关,剪切力增加与静脉内向重塑和AVF成熟障碍相关。
AVF动脉剪切力变化与静脉不尽相同。28例患者AVF术前桡动脉平均剪切力6 dyne/cm2,术后动脉直径和流量逐渐增加,剪切力亦升高,术后100d剪切力达39 dyne/cm2[18]。
AVF成熟后的吻合口剪切力约36 dyne/cm2,距吻合口25 mm以上剪切力基本恢复正常,内瘘静脉约10 dyne/cm2、供血动脉约25 dyne/cm2[19]。剪切力高于30 dyne/cm2启动血管损伤,>75 dyne/cm2激活白细胞黏附,>100 dyne/cm2血小板聚集和血栓形成,>350 dyne/cm2直接造成内皮细胞物理损伤、血小板黏附[15]。Ene-Iordache等[15]应用计算机模拟AVF血流动力学,发现低剪切力区域与内膜异常增生位置基本吻合。AVF吻合口根部静脉内侧壁、吻合口旁静脉内侧壁是血流紊乱、低剪切力区域,也是内膜异常增生的部位。环向力是血管环状面承受的张力,由血管壁产生,与血管内径、管壁厚度及血流量相关。理论上随着血管重塑,静脉内径增大、管壁增厚,环向力会逐步向术前基线水平回落。环向力增高会引起平滑肌细胞激活、增殖,细胞外基质成分增多,主要影响静脉壁中膜厚度。动脉环向力变化与静脉不尽相同。16例ESRD患者 AVF术后12周前桡动脉环向力呈增高趋势,管腔增大、管壁变薄,52周后环向力小幅下降,管壁增厚;而肱动脉环向力在术后52周内持续增高且管壁持续变薄[21]。
血管通路功能障碍的分子生物学机制
内膜增生是血管修复的主要病理生理过程,血管外膜在内膜增生过程中发挥重要作用。血管内皮细胞损伤后血管外膜成纤维细胞活化、增殖,并通过中膜迁移至内膜,在基质金属蛋白酶(MMP)、生长因子和细胞因子调控下转分化为肌成纤维细胞和血管平滑肌细胞;肌成纤维细胞和血管平滑肌细胞在中膜和内膜增殖,并合成细胞外基质,形成中膜增厚和内膜增生[1]。
内皮细胞损伤是静脉内膜增生的始发因素。尿毒症内环境,手术创伤引起血管痉挛和缺血,静脉-动脉吻合后血流动力学改变,缝线生物不相容,透析穿刺造成血管损伤,球囊扩张术均可引起血管内皮损伤。血管向外扩张重塑与内皮细胞依赖性血管反应相关,内皮细胞功能和一氧化氮(NO)在AVF成熟过程中发挥重要作用。内皮细胞持续损伤,诱导趋化因子、黏附因子和细胞因子生成增加,继而引发炎症反应,导致白细胞黏附、血小板激活、血管平滑肌细胞增殖和细胞外基质沉积[23]。NO是一种高效的血管扩张剂,高剪切力上调内皮细胞一氧化氮合酶(NOS)表达,NO合成和释放增加,引起血管扩张。内皮细胞暴露于15 dyne/cm2的剪切力15 min,NO释放量较剪切力为0时增加2倍[24]。NO与过氧化物生成过氧化亚硝酸盐,上调MMP表达,MMP可分解纤维蛋白和胶原蛋白,促进血管舒张,引起外膜肌成纤维细胞迁移至中膜和内膜,并增殖、分化和生成细胞外基质,在血管向内收缩和向外重塑中起重要作用。尿毒症毒素会引起内皮细胞功能障碍及NO活性降低。NO分泌不足会引起血管扩张障碍,过氧化物缺乏底物无法合成过氧化亚硝酸盐,影响血管重塑,过剩的过氧化物损伤血管内皮。非对称性二甲基精氨酸(ADAM)是一种内源性NOS抑制剂,减少NO生成,引起血管内皮功能障碍。ADAM为小分子物质,蛋白结合率较高,血液透析清除率低,尿毒症患者体内ADAM蓄积。研究发现AVF术前ADAM水平与内瘘早期血栓形成和狭窄无关[25],但其高水平与PTA术后AVF症状性再狭窄有关[26]。
在肾大部切除+AVF大鼠模型中,内皮型和诱导型NOS表达上调,应用非特异性NOS抑制剂N-硝基-L-精氨酸甲酯基精氨酸后,单核细胞趋化因子1(MCP-1)表达增强,瘘静脉内膜增生明显,内瘘血流量减少[27]。
氧化应激反应在AVF成熟早期明显增加,缺氧诱导因子-1,及其靶基因血红素加氧酶1(HO-1)表达增加[28]。HO-1是血红素分解代谢过程中的限速酶,在血管壁中具有强大的抗增殖和抗炎作用。HO-1基因敲除小鼠AVF模型,MCP-1、MMP-2和MMP-9表达显著增加,静脉内膜增生明显,导致AVF功能障碍;而野生型小鼠HO-1在瘘静脉表达增加,转化生长因子β(TGF-β)、血小板衍生生长因子(PDGF)、血管内皮生长因子和胰岛素样生长因子在两者表达相似[29]。通过腺病毒予小鼠AVF模型转染HO-1基因后,HO-1表达显著增加、活性上调,瘘静脉血流量增加、静脉壁厚度减少[30]。HO-1在非生理性剪切力状态下可提供血管保护作用[31]。HO-1缺乏诱导促炎和促氧化因子表达增加,可能是血管内膜异常增生、AVF功能障碍的重要因素之一。
MCP-1通过诱导单核巨噬细胞向炎症处聚集、内皮细胞活化、平滑肌细胞增殖和迁移,在动脉粥样硬化和其他血管疾病中发挥重要作用。小鼠模型AVF术后1周静脉段MCP-1 mRNA和MCP-1表达增加,伴随转录因子NF-κB和AP-1表达增加,MCP-1主要表达于内瘘静脉内皮细胞、增殖平滑肌细胞和浸润白细胞;而MCP-1基因敲除小鼠模型AVF术后6周内瘘通畅率明显升高、静脉壁厚度减少、静脉直径增大[32]。血管成形术后2天MCP-1增加超过基线水平25%是AVF再狭窄的独立预测因子[33]。内皮细胞损伤导致TGF-β1局部高表达。TGF-β1促进成纤维细胞增殖,诱导其表达α-平滑肌肌动蛋白,转分化为肌成纤维细胞,蛋白聚糖、胶原蛋白、透明质酸等细胞外基质合成增加;并介导单核细胞和中性粒细胞趋化至损伤部位,诱导单核细胞向巨噬细胞转化,刺激巨噬细胞释放TGF-β1、PDGF、成纤维细胞生长因子等具有促进成纤维细胞增殖和胶原蛋白合成作用的炎症因子。TGF-β1与AVF内膜增生和狭窄程度相关[34]。糜蛋白酶是贮存在肥大细胞的分泌颗粒,是已知最强的血管紧张素Ⅰ转换酶,增加TGF-β1表达和活化。血管紧张素Ⅱ和TGF-β1是刺激血管内膜增生的重要因素。CKD5期和ESRD患者静脉内膜和中膜糜蛋白酶表达增加。CKD5期患者血糜蛋白酶水平呈双峰分布,血浓度高者静脉内膜表达亦高,而ESRD患者血糜蛋白酶水平偏低,糜蛋白酶不能被透析器清除,可能与透析后使用ACEI/ARB制剂有关[35]。动物实验证实抑制糜蛋白酶可以减轻AVF、球囊扩张术后静脉内膜增生[34]。
目前与血管通路相关的基因研究主要集中在凝血级联反应、炎症反应以及内皮功能障碍。HO-1、MMP、TGF-β1、血管紧张素转换酶、血管紧张素Ⅱ受体1、低密度脂蛋白受体相关蛋白1、凝血因子V和纤溶酶原激活物抑制剂I等基因多态性可能是AVF功能障碍的潜在遗传危险因素[36]。MicroRNAs(miR)参与血管炎性反应、血管内膜增生、血管平滑肌细胞表型转化的调控。miR-21、miR-130a和miR-221在AVF狭窄静脉段中表达上调,主要分布在静脉增生内膜,而miR-133和miR-145表达下调;血miR-21水平升高是AVF狭窄的独立危险因素[37]。
综上所述,自体动静脉内瘘功能障碍主要原因是静脉内膜异常增生和血管外向扩张重塑障碍引起的内瘘静脉狭窄。低剪切力和高剪切力均会影响血管重塑。MCP-1、TGF-β、MMP、过氧化亚硝酸盐等炎症和氧化应激介质,以及基因多态性在成纤维细胞、肌成纤维细胞和平滑肌细胞活化、增殖和迁移中起重要作用。HO-1、NO可调控氧化应激和炎症反应、改善内皮功能(图4)。血流动力学、氧化应激、炎症反应、内皮细胞和基因等因素调控动静脉内瘘重塑,通过干预上述靶点有望促进内瘘成熟,延长内瘘寿命。
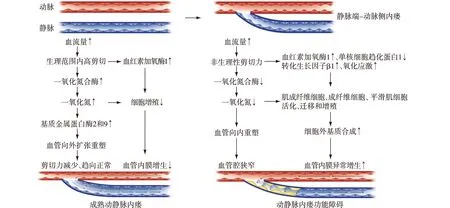
图4 动静脉内瘘成熟和功能障碍的病理生理机制
猜你喜欢
医院管理论坛(2022年7期)2022-10-14
橡塑技术与装备(2022年10期)2022-10-03
中国现代药物应用(2022年9期)2022-07-09
水利科技与经济(2021年11期)2021-12-04
世界最新医学信息文摘(2021年12期)2021-06-09
中西医结合心血管病杂志(电子版)(2020年22期)2020-12-09
山东医药(2020年20期)2020-08-06
中国循证心血管医学杂志(2017年5期)2017-07-01
浙江大学学报(工学版)(2016年2期)2016-06-05
中成药(2016年8期)2016-05-17
