
(2S,3S)-鞘胺醇的合成
2019-10-22 11:54:36宋艳玲杜新春仝瑞杰
沈阳化工大学学报 2019年3期
宋艳玲, 杜新春, 仝瑞杰
(沈阳化工大学 制药与生物工程学院, 辽宁 沈阳 110142)
鞘脂类化合物以鞘糖脂、鞘磷脂、1-磷酸-鞘胺醇和神经酰胺等形式广泛存在于动物、植物和真菌中.这类化合物作为细胞质膜的重要组成,不仅维持着细胞的基本结构,而且在细胞黏附、生长、分化、增殖和信号转导等基本活动中发挥着重要作用,具有抗微生物、抗肿瘤、免疫调节和似神经生长因子等生物活性[1-2].鞘胺醇(Sphingosine)是鞘脂类化合物的基本骨架,其分子结构由不饱和脂肪烃链和端位的2-氨基-1,3-二羟基两部分组成,如下所示:

目前,天然来源的鞘脂类化合物由于数量有限、提取困难、纯度较低等问题限制了其研究进展.因此,人工合成鞘脂类化合物显得尤为必要,特别是研究鞘脂类化合物的基本骨架鞘氨醇的合成方法具有重要的意义.已有文献报道的鞘氨醇的合成方法[3-6]绝大部分是采用氨基酸作为天然手性源以获得鞘氨醇的立体结构,而利用不对称合成方法得到手性中心的报道较少,且多为(2S,3R)-鞘胺醇的合成,关于(2S,3S)-鞘胺醇的合成报道甚少.本研究报道了通过不对称合成法合成(2S,3S)-鞘胺醇的新路线,如下所示:
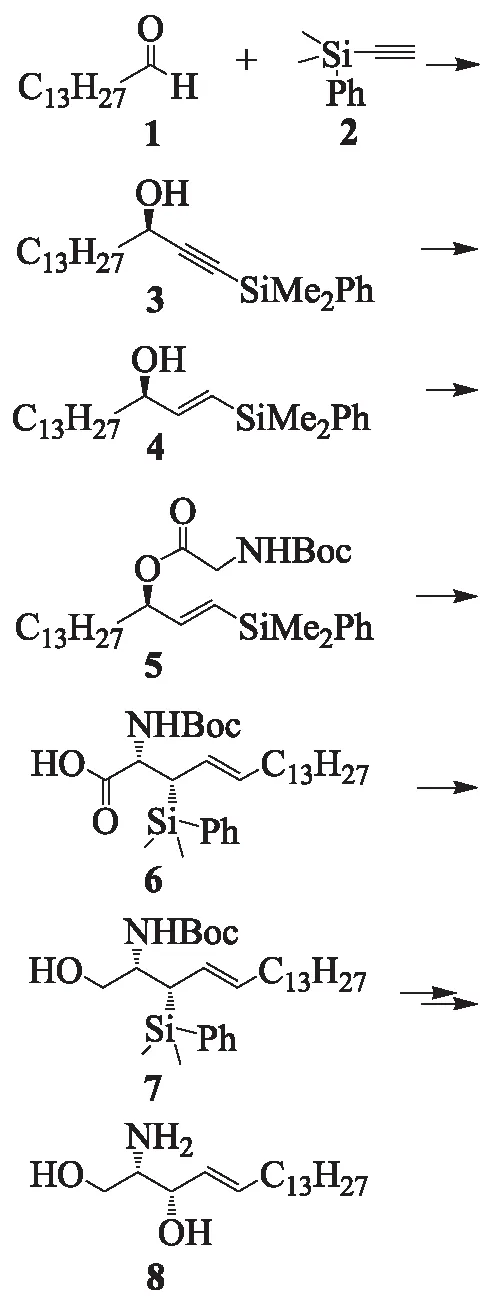
以肉豆蔻醛(1)和二甲基苯基硅烷基乙炔(2)为起始原料,在立体选择性催化剂(+)-N-甲基麻黄碱[(+)NME]的作用下经Carriera反应得到化合物(3);化合物(3)在Red-Al作用下发生立体选择性还原反应得到化合物(4);化合物(4)与叔丁氧基羰基保护的甘氨酸发生酯化反应,得到化合物(5);化合物(5)通过关键的Claisen重排反应得到化合物(6),再经过四氢铝锂还原反应得到化合物(7);最后经过氧化脱硅基反应和同时脱保护基反应得到目标产物(2S,3S)-鞘胺醇(8).该路线简便且产品光学纯度较高.
1 仪器与试剂
核磁共振用Bruker ARX-300测定;质谱采用LCQ Advantage MAX 10离子肼质谱仪测定;旋光度利用钠光(D line 589.3 nm)测量.反应通过Merck 60 F-254 硅胶薄层色谱板TLC检测,色谱柱填充硅胶为Merck 60(230-400 mesh).所用试剂均为市售分析纯.所有反应均在N2保护下进行.
2 实验方法
2.1 (R)-1-(二甲基苯基硅烷基)-1-十六炔-3-醇(3)的制备
将(+)NME(1.37 g,7.6 mmol)和三氟甲磺酸锌(2.26 g,6.2 mmol)加到密封管中,加入三乙胺(1.56 g,15.3 mmol)和甲苯(100 mL),室温下搅拌3 h,加入二甲基苯基硅烷(10.10 g,6.3 mmol),搅拌30 min,加入肉豆蔻醛(1.06 g,5 mmol).密封管加热至80 ℃,继续反应24 h.加入饱和的NH4Cl水溶液,淬灭反应.CH2Cl2萃取反应液,合并提取液,水洗,无水MgSO4干燥.抽滤,减压蒸馏,残留物经硅胶柱纯化[V(正己烷)∶V(乙酸乙酯)=20∶1],得到油状化合物1.59 g,收率86 %.1H-NMR(CDCl3,300 MHz),δ:0.418(s,6H),0.882(t,J=6 Hz,3H),1.259(brs,20H),1.438~1.494(m,2H),1.765~1.711(m,2H),4.400(t,J=6.6 Hz,1H),7.353~7.636(m,5H).
2.2 (R,E)-1-(二甲基苯基硅烷基)-1-十六烯-3-醇(4) 的制备
将红铝(质量分数65 %的甲苯溶液 0.41 mL,1.37 mmol)溶于2.0 mL乙醚中,反应液冷却至0 ℃,将化合物3(0.30 g,0.8 mmol)的1.0 mL THF溶液缓慢加到上述溶液中,温度升至室温,继续搅拌1 h.将反应液冷却至0 ℃,缓慢加入1 mol/L HCl终止反应,乙醚萃取反应液,合并提取液,水洗,无水MgSO4干燥.抽滤,减压蒸馏,残留物经柱层析纯化[V(正己烷)∶V(乙酸乙酯)=10∶1],得到油状化合物0.24 g.收率80 %.1H-NMR(CDCl3,300 MHz),δ:0.348(s,6H),0.882(t,J=6 Hz,3H),1.258~1.420(brs,20H),1.492~1.549(m,4H),4.113~4.134(dd,J=6.3 Hz,1H),5.939~6.005(dd,J=0.6,18.9 Hz,1H),6.093~6.173(dd,J=5.4,18.6 Hz,1H),7.341~7.363(m,3H),7.498~7.530(m,2H).
2.3 (R,E)-1-(二甲基苯基硅烷基)-3-(N-叔丁氧甲酰氨基)乙羰氧基-1-十六烯(5) 的制备
将(R,E)-1-(二甲基苯基硅烷基)-1-十六烯-3-醇(0.17g,0.45 mmol)溶于10 mL CH2Cl2中加入催化量的4-二甲氨基吡啶(DMAP,10 mg),室温搅拌30 min,冷却至0 ℃,将二环己基碳二亚胺(DCC,111.7 g,0.54 mmol)加到反应液中继续搅拌30 min,N-Boc-甘氨酸(44.7 mg,0.54 mmol)的CH2Cl2溶液加到反应液中,温度升至室温,继续搅拌过夜.减压蒸馏,残留物经柱层析纯化[V(正己烷)∶V(乙酸乙酯)=15∶1],得到化合物 0.23 g.收率95 %.1H-NMR(CDCl3,300 MHz),δ:0.410(s,6H),0.882(t,J=6 Hz,3H),1.258~1.420(brs,20H),1.386(s,9H),1.492~1.549(m,4H),3.931~3.941(d,J=3.0 Hz,2H),5.452~5.496(t,J=6.6 Hz,1H),5.939~6.005(dd,J=0.6,18.9 Hz,1H),6.093~6.173(dd,J=5.4,18.6 Hz,1H),7.348~7.399(m,3H),7.583~7.615(m,2H).
2.4 (2S,3S,E)-2-(N-叔丁氧基甲酰)氨基-3-(二甲基苯基硅烷基)-4-十八烯酸(6) 的制备
将二异丙基氨基锂(LDA)溶液1.9 mL[0.476 mol/L四氢呋喃(THF)溶液,0.9 mmol]冷却至-78 ℃,化合物 5(0.227 g,0.427 mol)溶于10 mL四氢呋喃溶液,缓慢加到LDA溶液中.搅拌20min后,加入叔丁基二甲基氯硅烷 (TBSCl)2.6 mL(0.5 mol/L THF溶液).将反应液缓慢升至室温,继续反应3 h.用1mol/L硫酸氢钠溶液淬灭反应,乙酸乙酯提取,合并提取液,水洗,无水MgSO4干燥.抽滤,减压蒸馏,残留物经柱层析纯化[V(正己烷)∶V(乙酸乙酯)=5∶1],得化合物150 mg,收率67 %.1H-NMR(CDCl3,300 MHz),δ:0.065(s,9H),0.876(t,J=6.6 Hz,3H),1.281(brs,20H),1.466(s,9H),1.836~2.008(m,2H),3.348(t,1H),4.948~4.980(d,J=9.6 Hz,1H),5.252~5.219(d,J=9.9 Hz,1H),5.384~5.456(dd,J=6.9,14.6 Hz,1H),7.260~7.376(m,3H),7.472~7.593(m,2H).
2.5 (2S,3S,E)-1-羟基-2-(N-叔丁氧基甲酰)氨基-3-(二甲基苯基硅烷基)-4-十八烯(7)的制备
将(2S,3S,E)-2-(N-叔丁氧基甲酰)氨基-3-(二甲基苯基硅烷基)-4-烯-十八酸(146.8 mg,0.28 mmol)溶入10 mL THF中,冷却至0 ℃,将四氢铝锂(1.0 mol/L THF溶液,0.83 mL)缓慢加至上述反应液中.加毕,温度升至室温,继续反应3 h.用10 mL乙酸乙酯稀释反应液,冷却至0 ℃,用体积分数5 %的HCl淬灭反应,乙酸乙酯提取,合并提取液,水洗,无水MgSO4干燥.抽滤,减压蒸馏,残留物经柱层析纯化[V(正己烷)∶V(乙酸乙酯)=5∶1],得到化合物135.3 mg,收率95 %.1H-NMR(CDCl3,300 MHz),δ:0.339(s,6H),0.857(t,J=6.6 Hz,3H),1.235(brs,20H),1.406(s,9H),1.872(m,2H),1.947~1.981(m,2H),3.441~3.500(dd,J=6.3,11.4 Hz,1H),3.629~3.677(dd,J=3,11.4 Hz,1H),3.758(brs,1H),5.208~5.242(d,J=10.2 Hz,1H),5.304~5.376(dd,J=8.4,14.7 Hz,1H),7.252~7.370(m,3H),7.472~7.513(m,2H).
2.6 (2S,3S)-鞘胺醇(8)的制备
将(2S,3S,E)-1-羟基-2-(N-叔丁氧基甲酰)氨基-3-(二甲基苯基硅烷基)-4-十八烯(135.3 mg,0.27 mmol)溶入5 mL三氟乙酸和冰醋酸(体积比1∶1)的混合溶剂中,加入三氟乙酸汞(122.2 mg,0.28 mmol),室温下搅拌1 h,然后加入过氧乙酸(60 mg,0.78 mmol),室温下继续搅拌12 h.用乙酸乙酯10 mL稀释反应液,依次用饱和的碳酸氢钠溶液、饱和的亚硫酸钠溶液和水洗,无水MgSO4干燥.抽滤,减压蒸馏,残留物经柱层析纯化[V(正己烷)∶V(乙酸乙酯)=5∶1],得到化合物67 mg,收率85 %.1H-NMR(CDCl3,300 MHz),δ:0.816(t,J=6.9 Hz,3H),1.235(brs,20H),1.406(s,9H),1.872(m,2H),1.964~2.032(dd,J=6.9,14.1 Hz,2H),2.729~2.783 3(dd,J=4.5,6.0 Hz,2H).3.527~3.643(m,2H),3.629~3.677(dd,J=3,11.4 Hz,1H),3.838(s,4H),3.983(s,1H),5.343~5.418(dd,J=7.5,14.1 Hz,1H),5.650~5.725(m,J=8.2,15.3 Hz,1H); MS(m/z):299.28[M+H]+; [α]25D-25.3(c0.30,氯仿).
3 结果与讨论
以不对称合成方法建立了C(2),C(3)的立体结构及一个C(4)位的E型双键结构.缩短了反应步骤,提高了反应总收率,并且对多步反应条件进行优化与改进.
在化合物3的合成过程中采用(+)-N-甲基麻黄碱作为立体选择性催化剂的Carriera反应.根据文献报道的Carriera反应条件[7],按n(化合物1)∶n(化合物2)∶n[(+)NME]∶n(三氟甲磺酸锌)∶n(三乙胺)=1∶1∶1.1∶1.2∶1.2的投料比例室温反应48 h,起始原料没有变化,无新的生成产物.通过改进反应条件,将投料比例提高为n(化合物1)∶n(化合物2)∶n[(+)NME]∶n(三氟甲磺酸锌)∶n(三乙胺)1∶10 ∶1.5∶1.2∶3,同时提高反应温度至65 ℃,采用密封管中加压反应20 h,反应得以顺利进行,反应收率提高至85 %.
在化合物4的合成过程中尝试不同的选择性还原反应.实验结果表明,在红铝还原反应条件下可得到较高收率的E构型还原产物,结果如表1所示.

表1 不同还原反应对结果的影响
Claisen重排反应是合成过程中最重要,也是最难控制的一步反应.对比了LDA、KHMDS和LHMDS等金属碱对实验的影响;对螯合剂ZnCl2、TMSCl和TBSCl进行了研究;同时对反应温度和时间进行研究.确定了最优化的Claisen重排反应条件:3倍量的TBSCl 和2倍量的LDA,-78 ℃至室温条件下反应2 h,反应收率为65 %.
化合物7的氧化反应过程中,将其溶入冰醋酸和三氟乙酸(体积比1∶1)的混合溶剂中,加入三氟乙酸汞,室温下搅拌0.5 h,然后加入过氧乙酸,发生氧化脱硅基反应,同时该反应条件下会发生脱Boc保护反应,因此,确定了一勺烩的反应条件,得到最终目标产物(2S,3S)-鞘胺醇.
4 结 论
鞘胺醇是鞘脂类化合物的基本骨架,关于(2S,3S)-鞘胺醇的报道非常少,本文对其合成路线进行研究,成功制备出了目标化合物,各项指标均与文献报道值相符.因此,该合成路线可应用于人工合成鞘脂类化合物的进一步研究.
猜你喜欢
上海计量测试(2020年1期)2020-03-18 02:31:22
农药科学与管理(2019年8期)2019-11-23 08:04:44
西南石油大学学报(自然科学版)(2018年6期)2018-12-26 01:00:18
浙江大学学报(工学版)(2016年2期)2016-06-05 09:20:51
环境科技(2015年1期)2015-11-08 12:10:50
化学工业与工程(2015年1期)2015-02-10 03:01:33
食品工业科技(2014年15期)2014-03-11 18:17:49
无机化学学报(2014年12期)2014-02-28 17:34:01
无机化学学报(2014年7期)2014-02-28 17:32:28
中国洗涤用品工业(2012年4期)2012-03-20 15:39:34
