
Lesson Ninety-one Drug-induced fatal arrhythmias:Acquired Brugada syndromes
2019-10-17 07:23:38
心电与循环 2019年5期
Current concept of Brugada syndrome
Brugada syndrome(BrS)is recognized by the characteristic coved-type ST elevations(denoted as type 1)in right precordial leads in the absence of structural heart disease.Fig.1 depicts typical ECG features induced by pilsicainide tolerance test.According to the J-Wave Syndromes Expert Consensus Report published in 2016,BrS is diagnosed by the following criteria:
1.In patients with spontaneous type 1 ST-segment elevation of 2 mm or more in at least one lead among leads V1-V3,positioned in the 2nd,3rd or 4th intercostal space;or,
2.In patientswith drug-induced type 1 ST-segment elevation of 2 mm or more in at least one lead among leads V1-V3,positioned in the 2nd,3rd or 4th intercostal space and at least one of the following:(i) unexplained sudden cardiac death (SCD) or documented ventricular fibrillation(VF)/polymorphic ventricular tachycardia (VT), (ii) nocturnal agonal respirations,(iii)syncope of probable arrhythmic cause,(iv)first or second degree relative with definite BrS.
In 1992,BrS was presented as a genetic disorder showing familial aggregation that was associated with a characteristic ECG pattern and sudden cardiac death secondary to polymorphic VT or VF.Mechanisms behind creation of macroscopic arrhythmias by cellular currentchangesremain controversial.Mainly two hypotheses,based on repolarization or depolarization changes,have been proposed and might be acting alone orin conjunction.The firsthypothesis proposes amplification of the inherent differences in repolarization patterns ofthe differentlayers of myocardium which is commonly referred to as the spatial dispersion of repolarization(SDR).Transient outward current(Ito) is the most important current ascribed to SDR.It is present in the epicardial cells and M cells,but not in the innermost of the three myocardial layers,the endocardium (Fig.2A).Moreover,in the right ventricle,Ito is of nearly three fold magnitude compared to the left ventricle which is behind the arrhythmogenesis of right ventricular origin in BrS.
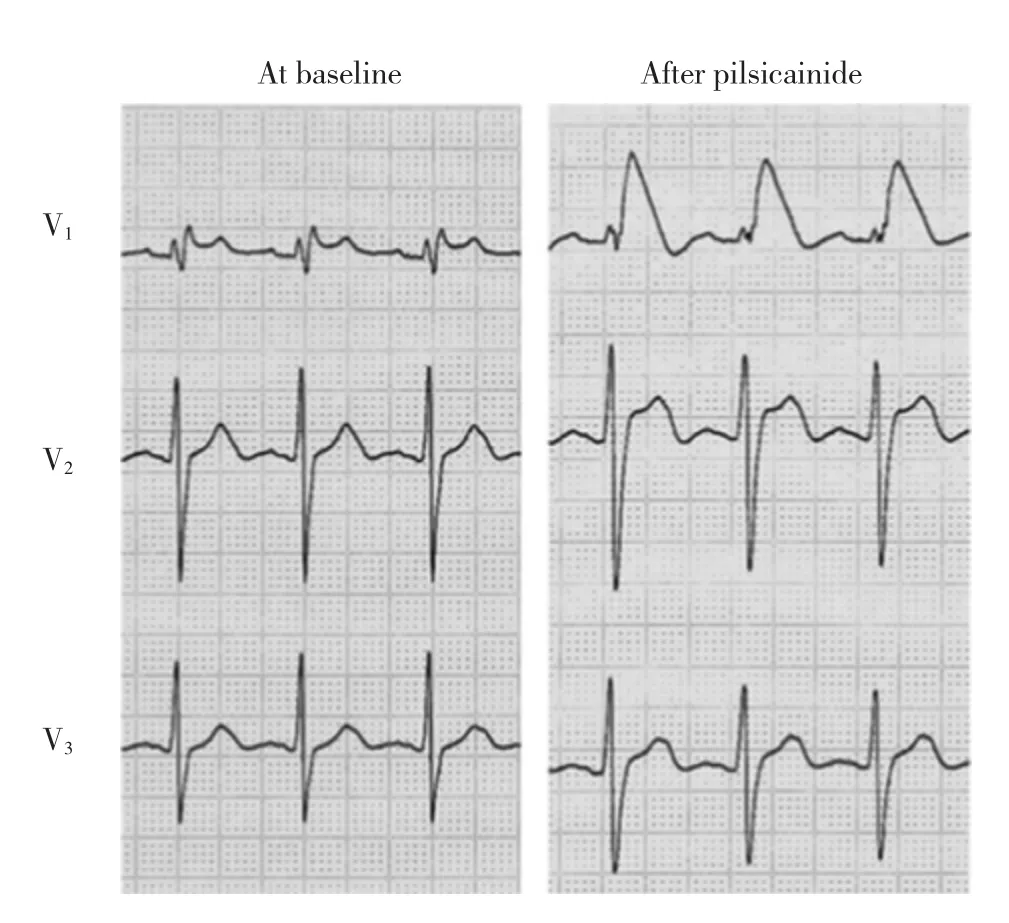
Fig.1 Pilsicainide-induced Brugada pattern ECG changes:the left panel shows baseline precordial leads and the right panel shows the same after intravenous injection of pilsicainide(0.8mg/kg)
Transmuralvoltage difference in phase 1 repolarization caused by heterogeneous expression ofIto between endocardium and epicardium explains the abnormal J point elevation seen in BrS(Fig.2B).Ito prominence subjects cells to all-or-none repolarization once the nadir of action potentials(AP)phase 1 reaches below the activation threshold of L-type calcium channels resulting in loss of the AP dome and shortening of the AP duration (Fig.2C).This is contributed by certain pathophysiologic conditions and drug interventions that decrease depolarizing currents.Heterogeneous loss of the AP dome in the epicardium results in epicardial dispersion of repolarization and contributes more to the inherent transmural heterogeneity.When the dome of AP is lost in epicardial cells,but not in endocardial cells,a transmural voltage gradient between endocardium and epicardium occurs,showing itselfas the characteristic ST segment elevations inscribed on the ECG.
Progression of currents,within the epicardium or transmurally,from areas where phase 2 is preserved to those where it is lost,cause short coupled re-excitations named phase 2 re-entry which provides the trigger for VT and VF seen in BrS(Fig.2D).This hypothesis is also supported by the ECG response of BrS patients to autonomic changes,i.e.parasympathetic activation induced increase inIto and/or decrease inICa attenuate the spike and dome pattern of the AP increasing the dispersion of repolarization and accentuating the ST elevation seen on ECG.
A second hypothesis for development of arrhythmias in BrS proposes slowing of depolarization and conduction by fibrosis and decreasedINa in right ventricular outflow tract(RVOT) as the primary mechanism.Relatively delayed depolarization in RVOT with respect to other sites of RV creates regional potential differences that ascribe itself as the ST-segment elevation seen on the right precordial leads.

Fig.2 Dispersion ofrepolarization and phase2 reentry hypothesis in Brugada syndrome:A)Ito is themain current responsible for the phase 1 of the AP and is absent in endocardial cells.B)Transmural voltage difference in phase 1 repolarization caused by heterogeneous expression of Ito between endocardium and epicardium explains the abnormalJ point elevation seen in J wave syndromes. C)Ito prominence and/or decrease in inward sodium or calcium currents in BrS subject cells to all-or-none repolarization once the nadir of AP phase 1 reaches belowthe activation threshold ofL-type calcium channels thereby resulting in loss of the AP dome and shortening of the AP duration.D)Progression of currents,within the epicardiumor transmurally,from areas where phase 2 is preserved to those where it is lost cause short coupled re-excitations named phase 2 re-entry which provides the trigger for VT and VF seen in BrS.
Drug-induced Brugada syndrome
In biologically predisposed patients,environmental factors could modulate the observed phenotype.Autonomic changes,hormonal changes,alcohol,fever,and medications can turn a normal ECG to a BrS ECG.Several studies reported that exercise,atropine and isoproterenol infusion could normalize a BrS ECG,whereas muscarinic and selective α-adrenoceptor stimulation,through reflexvagalactivation,could augment the ST elevation.This also explains the propensity of VT/VF episodes during night time in BrS.Similarly,while targeting sympathetic activity via left cardiac sympathetic denervation is a widely accepted therapy for intractable long QT syndrome,it may increase arrhythmias in BrS.Several studies reported successful prevention of intractable episodes of VF in BrS using phosphodiesterase inhibitors,milrinone and cilostazol.These drugs elevate cyclic AMP levels in the cell and increase L-type calcium current(ICa),thereby restoring the epicardial AP dome.
Fever has long been recognized as a risk factor for arrhythmias seen in BrS.Some SCN5A mutations cause alteration in sodium channel kinetics only at high temperatures.Other studies have suggested that sensibility to fever may not be due to mutations,but due to temperature-dependent properties of the wild-type SCN5A orIto,as well as fever induced facilitation of spontaneous activity in RVOT and Purkinje fibers.
As can be summarized from above discussions,any drug that would decreaseINa orICa or increaseIto would be arrhythmogenic in BrS.According to their mechanism of action,these drugs can be classified into three major groups:
Sodium channel blockers
The amplitude ofINa underlies the maximum positive membrane potential reached at the end of AP phase 0.IfINa is diminished,then AP phase 1 starts at lower membrane potential and ends at more negative potentials.The balance of inward and outward currents at the end of phase 1 determines whether or notICa can overcome the outward currents and induce formation of the AP dome.Since endocardium and epicardium have different responses toINa blocking agents,they cause transmural heterogeneity in the formation of the AP dome.Class IA and IC antiarrhythmic agents are therefore utilized in unmasking the BrS phenotype.
Tricyclic antidepressants(amitriptyline,desimipramine,nortriptyline,clomipramine,imipramine)and tetracyclicantidepressants(maprotiline),viatheirsodium channel blocking effects,are known to precipitate Brugada type ECG by diminishing the net inward current at the end of phase 1 AP.
Neuroleptic drug class phenothiazines(chlorpromazine,trifluoperazine,cyamemazine,perphenazine),haloperidol and loxapine can induce BrS type ECG via sodium channel blocking properties.
SSRIs,particularly fluoxetine,have sodium and calcium channel blocking properties in mammalian ventricular myocytes and have been shown to induce BrS type ECG.Another SSRI,citalopram,has been found to inhibit activation ofINa as well.Other SSRIs,fluvoxamine and paroxetine are also reported to unmask BrS ECG.
Antihistaminic drug,terfenadine,which has bothINa andICa blocking properties was shown to be more effective in provoking VT thanINa blockers alone in canine models of ventricular wedge preparations.Among sodium channel blocking agents those with potentIto blocking effects are less likely(flecainide and disopyramide)or even not likely(quinidine)to induce arrhythmogenesis.Lithium,cocaine,cannabis,alcohol and anesthetic agents(propofol,bupivacaine,ketamine)also unmask BrS phenotype by decreasingINa.Even though methadone is better known for its association with drug-induced long QT Syndrome,it has also been reported to induce BrS ECG.Another synthetic opioid tramadol which blocks neuronal sodium channels is also reported to unmask BrS type ECG.Many antiepileptic medications assert their affect via blockage of the neuronal sodium channels.Some of these have also been shown to alter cardiac sodium channel kinetics,while some are yet to be shown to do so.
Patients who develop a syncopal episode and found in a"postictal state"1could have had an arrhythmic episode and found in a state of confusion due to cerebral hypoperfusion which could be mistaken for postictal status.In these patients well-intended therapeutic maneuvers with sodium channel blocking antiepileptic medications(phenytoin,carbamazepine,primidone,lamotrigine,and topiramate etc.)could induce BrS and have deleterious consequences.Therefore,diagnosis of seizure disorder should only be made after careful exclusion of the possibility of BrS.
Potassium channel openers
The balance of inward and outward currents at the end ofAP phase1 determineswhetherornot repolarization will occur in an all-or-none fashion;therefore,any increase in outward currents(IKATP,Ito,andIKr)can cause loss of the AP dome and result in a vulnerable window in voltage gradients between areas where these currentsare differentially expressed.IKATP activators,pinacidil and nicorandil,have been shown to cause loss of the AP dome in areas whereIto is prominent with resultant phase-2 reentry in experimental models.Glucose and insulin could unmask the BrS phenotype by decreasing serum potassium concentrations and accentuatingIto.Diuretics and distal renal tubular acidosis can act in a similar manner by decreasing serum potassium levels.
Calcium channel blockers
ICa is the main current to form the AP dome.If it is diminished and cannot overcome the outward balance of currents at the end of phase 1 AP,the dome disappears.It has been shown thatIto which is the main current responsible for formation of phase 1 is heterogeneously expressed in between different layers of the myocardium,therefore a strongICa acts as the rescuer of the AP dome whereIto is increased.The basis of provocation of BrS phenotype with calcium channel blockers,in part,is due to failing to reach the potential threshold needed to rescue the AP dome in areas whereIto is strong(RVOT).Calcium channel blockers and β-blockers unmask BrS by decreasing L-type calcium currents.Acetylcholine,by inhibition ofICa,can result in loss of the AP dome and trigger arrhythmias.Nitrates,similarly,have L-type Ca channel blocking properties and could induce BrS phenotype. Of theINa blockers listed above,trifluoperazine,cyamemazine,fluoxetine,terfenadine,ketamine,alcohol etc.also blockICa increasing the effects of these drugs in uncovering the BrS phenotype.
词 汇
agonal adj.痛苦的,濒死痛苦的,呻吟待毙的,临死前的
aggregation n.聚集,聚集体,聚合作用
macroscopic adj.肉眼可见的,宏观的
spatial adj.空间的
ascribe v.把…归因于,把…归咎于,认为是…的特点
innermost n.&adj.最深处;内心深处的,最靠近中心的,最深处的
heterogeneous adj.各种各样的,由很多种类组成的,不均质的
propensity n.倾向,癖好,习性
intractable adj.难驾驭的,难加工的,难治疗的
arrhythmogenesis n.致心律失常,心律失常
注 释
1.postictal state“发作后状态”,通常指癫痫发作过后的一种状态,时间是从癫痫发作结束到恢复基础(发作前)状态期间,表现为意识混乱、昏睡、精神异常、昏迷,也可出现暴力行为,记忆缺失等,确切的机制未完全明了。
参考译文
第91课 药物诱发的致命性心律失常-获得性Brugada综合征
Brugada综合征的现代概念
Brugada综合征(BrS)特征表现为右胸导联穹形ST段抬高(1型)而无结构性心脏疾病。图1显示吡西卡尼耐力试验诱发的典型心电图特征。根据2016 J-波综合征专家共识报告,BrS诊断标准如下:
1.电极位于第2,第3或第4肋间的V1~V3上,至少有一个导联上出现自发性1型ST段抬高2mm或以上;或
2.电极位于第2,第3或第4肋间的V1~V3上,至少有一个导联上出现药物诱发的1型ST段抬高2mm或以上,同时具备下列条件之一:(i)不能解释的心源性猝死或记录到心室颤动/多形性室性心动过速,(ii)夜间濒死样呼吸,(iii)晕厥可能由心律失常引起,(iv)一级或二级亲属中有明确的BrS。
1992年,BrS以遗传性家族聚集性疾病加以报道,伴随特征心电图表现和继发于多形性室性心动过速(VT)或心室颤动(VF)的心源性猝死。产生这种明显心律失常的细胞电流变化机制仍存争议。基于复极和除极变化,主要提出两种假说,单一或联合起作用。第一种假说推测不同心肌层之间的内在复极差异得到放大,通常称作复极空间离散(SDR)。短暂外向电流(Ito)是SDR中最重要的电流。出现于心外膜细胞和M细胞,而不出现于三层心肌中的最内层心内膜(图2A)。此外,右心室Ito量是左心室的近3倍,这是BrS右心室致心律失常的原因。
心内膜与心外膜之间Ito表达不均一导致的复极1相透壁电位差解释了BrS的异常J点抬高(图2B)。Ito明显而致细胞面临全-或-无复极(现象),一旦动作电位1相谷值到达L-型钙通道激动阈值以下时即导致动作电位穹顶消失和动作电位时程缩短(图2C)。某些降低复极电流的病理状态或药物干预促进这一现象。心外膜动作电位穹顶的不均一丢失导致心外膜复极离散而进一步加剧内在的透壁不均一性。当心外膜细胞丢失动作电位穹顶而心内膜不丢失时,心内膜与心外膜之间即发生透壁电压梯度,表现为心电图上的特征性ST段抬高。
心外膜内或透壁电流扩布,从保留动作电位2相的部位流向动作电位2相缺失的部位,引发短偶联间期的再兴奋,称作2相折返,这成为BrS VT和VF发作的触发机制(图2D)。这一假说得到BrS患者因自主神经张力改变而发生ECG变化的支持,即迷走神经激动引发的Ito增加和(或)ICa减少降低了动作电位的尖峰和穹顶图形,从而加剧复极离散和ECG ST段抬高。
第二种关于BrS心律失常的假说推测纤维化引起除极与传导延缓,右心室流出道(RVOT)INa减少是主要机制。相对于右心室其他部位,RVOT的相对延缓除极导致局部电位差,这在右胸导联上表现为ST段抬高。
药物诱发的Brugada综合征
对于生物学上易感患者,环境因素将改变观察到的表型。自主神经变化、激素变化、饮酒、发热和药物可以使正常心电图转变成BrS心电图。多个研究报道,运动、阿托品和异丙肾上腺素滴注可使BrS心电图正常化,而毒蕈碱和选择性α-肾上腺受体刺激,通过反射性迷走激活,可以加剧ST段抬高。这也解释了BrS患者倾向于夜间发生VT/VF。同样,虽然以交感神经激动为目标的左侧心脏神经离断术是广为接受的顽固性长QT间期综合征治疗方法,却增加BrS的心律失常。多个研究报道,应用磷酸二酯酶抑制剂米力农和西洛他唑能成功预防BrS的顽固性VF发作。这些药物提高细胞内环AMP水平,增加L-型钙电流(ICa),从而恢复心外膜动作电位穹顶。
一直认为发热是BrS心律失常的危险因素。有些SCN5A突变只能在高温下改变钠通道动力。其他研究提示对热的敏感性并非归因于突变,而是野型SCN5A或Ito的热依赖特性,以及发热助长了RVOT和浦肯野纤维的自发活动。
基于上述讨论总结,任何可降低INa或ICa或增加Ito的药物,均可引发BrS心律失常。根据它们的作用机制,这些药物可以分成三大组:
钠通道阻断剂
INa的幅度决定达到动作电位0相顶端的最大正向膜电位。如INa减少,动作电位1相起始膜电位较低,结束时负向电位更明显。1相结束时的内外电流平衡决定ICa能否克服外向电流而形成动作电位穹顶。因为心内膜与心外膜对INa阻断剂的反应不同,它们可以导致动作电位穹顶形成过程中的透壁不一致性。因此,IA和IC抗心律失常制剂用于揭示BrS表型。
已知三环类抗抑郁制剂(阿米替林,去甲丙咪嗪,去甲阿米替林,氯丙米嗪,丙咪嗪)和四环类抗抑郁制剂(马普替林),通过钠通道阻断作用,减少动作电位1相结束时净内向电流而促发Brugada心电图。
抑制神经药物吩噻嗪类(氯丙嗪,三氟拉嗪,氰美马嗪,奋乃静),氟哌丁苯和洛沙平可通过钠通道阻断特性而诱发BrS心电图。
选择性血清素重摄取抑制剂(SSRIs),特别是氟西汀具有哺乳类动物心室细胞的钠和钙通道阻断特性,已显示能诱发BrS心电图。另一SSRI西酞普兰也能抑制INa激活。其他SSRIs氟伏沙明和帕罗西汀也能揭示BrS心电图。
抗组胺类药物丁苯哌丁醇具有INa和ICa阻断特性,在犬契形标本上显示其激发VT的作用超过单一的INa阻断剂。在钠通道阻断剂中,那些具有明显Ito阻断作用的少有可能(氟卡尼和丙吡胺)或甚至不可能(奎尼丁)诱发心律失常。锂、可卡因、大麻、酒精和麻醉剂(丙泊酚,布比卡因,氯胺酮)也可通过减少INa而揭示BrS表型。尽管众所周知美沙酮与药物诱发长QT综合征相关,也有报道其可诱发BrS心电图。另一合成麻醉制剂曲马多阻断神经元钠通道,也有报道可揭示BrS心电图。许多抗癫痫药物通过阻断神经元钠通道而起作用。其中有些也改变心脏钠通道动力,虽然有些尚需证实。
发生晕厥并处于“发作后状态”的患者可能有心律失常发作,并出现脑低灌注引起的混乱状态,这可误判为发作后状态。对这些患者采用钠通道阻断剂抗癫痫药物(苯妥英,卡马西平,扑痫酮,拉莫三嗪,和托吡酯等)的定向治疗措施可诱发BrS和导致不良后果。因此,癫痫诊断只能在仔细排除BrS后才能得以确立。
钾通道开通剂
动作电位1相结束时内外向电流的平衡决定复极是否发生全-或-无形式,因此,任何外向电流(IKATP,Ito和IKr)的增加可引起动作电位穹顶的丢失,并导致电流表达不同区域之间电压梯度上的易损窗口。IKATP激动剂匹那地尔和尼可地尔已显示能导致实验模型上Ito为主导而引起2相折返区域的动作电位穹顶丢失。葡萄糖和胰岛素通过减少血清钾浓度和增加Ito而揭示BrS。利尿剂和远端肾小管酸中毒通过减少血清钾浓度而起到类似作用。
钙通道阻断剂
ICa是形成动作电位穹顶的主要电流。如在动作电位1相结束时ICa降低并且不能克服外向电流的平衡,穹顶就消失。已显示形成动作电位1相的主要电流在心肌的不同层之间表达不一致,因此,在Ito增加区域强ICa是动作电位穹顶的挽救者。钙通道阻断剂激发BrS的基础,部分源于难以达到所需的电位阈值来挽救Ito强大区域(RVOT)的动作电位穹顶。钙通道阻断剂和β-阻断剂通过降低L-型钙电流而揭示BrS。乙酰胆碱通过抑制ICa导致动作电位穹顶的丢失并促发心律失常。同样,硝酸盐具有L-型钙通道阻断特性,能引发BrS表型。在上述所列的INa阻断剂中,三氟拉嗪,氰美马嗪,氟西汀,丁苯哌丁醇,氯胺酮,酒精等也能阻断ICa而增加这些药物揭示BrS表型的效应。
图1吡西卡尼诱发的Brugada心电图变化:左图为基础状态胸导联心电图,右图为静脉注射吡西卡尼(0.8mg/kg)后胸导联心电图。
图2Brugada综合征复极离散和2相折返假说:A:Ito是动作电位1相的主要电流,心内膜细胞缺乏此电流;B:心内膜与心外膜之间不一致的Ito表达导致复极1相透壁电位差,是J波综合征中J点抬高的原因;C:BrSIto明显和(或)内向钠或钙电流减少致使细胞面临全-或-无现象,一旦动作电位1相谷值达到L-型钙通道激动阈值以下时即导致动作电位穹顶丢失和动作电位时程缩短;D:心外膜内或透壁电流扩布,从保留动作电位2相的部位流向动作电位2相缺失的部位,引发短偶联间期的再兴奋,称作2相折返,这成为BrS VT和VF发作的触发机制。
猜你喜欢
新医学(2024年6期)2024-07-13 07:36:17
国际呼吸杂志(2019年2期)2019-02-14 06:11:26
四川农业科技(2018年6期)2018-03-17 18:58:28
临床超声医学杂志(2017年7期)2017-08-10 23:37:24
温州医科大学学报(2016年1期)2016-04-06 02:11:54
江苏大学学报(医学版)(2015年5期)2015-04-16 05:30:07
中西医结合心血管病杂志(电子版)(2015年35期)2015-01-21 08:38:08
中国海洋大学学报(自然科学版)(2014年12期)2014-02-28 12:22:05
中国药理学通报(2013年11期)2013-12-08 06:58:14
中国医院用药评价与分析(2012年2期)2012-11-06 10:35:40
