
糖尿病周围神经病变及痛性糖尿病神经病变机制新方向*
2019-09-20 03:46:22陈旭辉张传汉
中国疼痛医学杂志 2019年9期
李 婷 陈旭辉 张 玥 张传汉
(华中科技大学同济医学院附属同济医院麻醉与疼痛学教研室,武汉430030)
糖尿病(diabetes mellitus, DM)是一组以高血糖为特征的代谢性疾病,常伴有复杂的代谢综合征。2007 至2008 年,全国14 个省市进行的糖尿病流行病学调查显示,我国20 岁以上成年人的糖尿病患病率为9.7%,糖尿病前期为15.5%,且发病人口仍在迅速增加[1]。DM 病人可能会出现严重的慢性并发症,糖尿病周围神经病变(diabetic peripheral neuropathy, DPN)是DM 最常见的并发症之一[2],常见的形式为糖尿病感觉运动多发性神经病(diabetic sensory motor polyneuropathy, DSPN),影响外周神经系统(peripheral nervous system, PNS)的感觉、运动和自主神经功能。其典型的表现为感觉神经病变,具有特征性的手套袜套分布[3],且50%左右的糖尿病和13%左右的糖耐量受损的病人伴随着神经病理性疼痛,导致病人生活质量和工作能力受损[1]。因此,更好的理解DPN的机制可以为早期诊断和治疗,防止远期并发症的发生提供依据[4]。
近年来,DPN 领域的研究集中在与神经元的代谢和/或氧化还原状态有关的DPN 相关途径,如多元醇途径,己糖胺途径,晚期糖基化终末产物堆积,神经元氧化应激等等,但这些临床研究成果尚未实现成功转化,临床试验并未成功,主要原因是药物无法到达周围神经或者药物的肝毒性较大而无法应用[4,5]。因此本综述总结了DPN 和PDPN 发病机制的最新方向,包括施万细胞与这些途径的相互作用,施万细胞和轴突之间强烈相互作用的结节区域、内质网应激对DPN 发病机制的贡献,以及Na+,Ca2+通道,小胶质细胞和巨噬细胞在PDPN 中的作用,为更好的理解DPN 和PDPN 提供参考。
一、DPN 的发病机制进展
1.施万细胞在DPN 的作用
糖尿病神经病变的大多数临床和基础研究都集中在高糖对神经元的影响上[3]。然而,越来越多的数据证实施万细胞在维持神经元结构和功能,滋养轴突及促进受损神经元存活和修复方面发挥着不可或缺的作用。糖代谢异常使得施万细胞神经毒性中间体聚集并减少神经营养因子的产生,最终导致轴突变性,内皮功能障碍和DPN[3]。
施万细胞是PNS中最丰富的细胞,包括两大类:髓鞘和无髓鞘施万细胞,一起包裹着周围神经的轴突。高糖状态下,施万细胞功能受到干扰,损害胶质、轴突通讯和神经稳态,导致纤维丢失,神经变性和疼痛[6]。在DPN 的病理过程中首先出现节段性轴突脱髓鞘和髓鞘再生继而出现轴突变性,这表明施万细胞受损可能是神经纤维损伤的基础,是DPN发病的第一步[6]。
(1)施万细胞中多元醇途径通量增加参与DPN:高糖驱动的多元醇途径流量增加是DPN 研究最多的致病机制。在神经内膜中,醛糖还原酶主要由髓鞘化施万细胞表达,因此,高糖主要通过醛糖还原酶介导的多元醇途径对施万细胞产生毒性。啮齿动物糖尿病模型中,施万细胞分泌的沙漠刺猬因子、睫状神经营养因子19、髓鞘蛋白,神经生长因子和神经营养因子-3 等减少,导致神经纤维进一步丢失和传导速度受损,促使DPN 的发展[6]。已有研究证实醛糖还原酶抑制剂(主要应用于施万细胞)可改善DSPN 模型的轴突病变和施万神经病变[2]。因此,高糖诱导的施万细胞中多元醇途径通量的增加可能是主要的致病机制之一[3]。
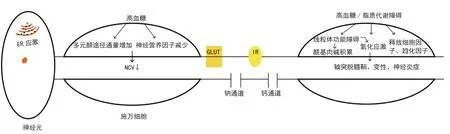
图1 DPN 的发病机制。持续高糖状态可导致施万细胞中的多元醇途径通量增加,髓鞘蛋白,CNTF,NGF和NT-3等神经营养因子的产生减少,线粒体功能障碍,氧化应激,以及炎症因子释放,导致神经炎症,轴突脱髓鞘变性,NCV减慢进而促进DPN。结节区域富集的GLUT 定位障碍影响能量供应,IR对胰岛素敏感性降低,进而促进了轴突变性;此外,该区域的钠通道,钙通道均参与疼痛性的糖尿病神经病变(PDPN)。神经元中的ER 应激参与周围神经的氧化应激和DPN 的发展。缩写:DPN:糖尿病周围神经病变;PDPN(疼痛性的糖尿病神经病变)ER:内质网;NCV:神经传导速度;GLUT:葡萄糖转运蛋白;IR:胰岛素受体。
(2)施万细胞的氧化应激和线粒体功能障碍加剧DPN:施万细胞中3-硝基酪氨酸和诱导型一氧化氮合酶水平增加,提示了高糖可以诱导施万细胞的亚硝化应激反应。体外高糖诱导Bcl-2 水平降低提示施万细胞线粒体应激[3]。这些应激反应导致酰基肉碱的积累,释放后诱导轴突变性。过量葡萄糖还可导致晚期糖基化终产物(advanced glycation end products, AGEs)形成,继而激活AGE 受体(receptor for AGE RAGE)并促进活性氧自由基(reactive oxygen species, ROS)形成。AGE 诱导的关键蛋白,脂质和核酸的修饰和氧化应激可改变施万细胞的结构和功能,对轴突产生不利影响,导致糖尿病神经病变加剧[3]。此外有研究表明,醛糖还原酶抑制剂可预防糖尿病大鼠神经中脂质和DNA 氧化损伤,表明施万细胞中的多元醇途径加剧了氧化应激[3]。
研究显示高糖还可以驱动施万细胞线粒体蛋白质的重塑,导致ATP 合成酶α 和β 亚基的表达增加,提高总体氧耗率导致呼吸能力不足[7],这表明高糖诱导施万细胞线粒体功能障碍,降低其氧化磷酸化的效率,进而使神经胶质细胞对轴突的支持受到干扰,依次导致无髓纤维、有髓纤维发生原发性神经元变性[2]。
(3)施万细胞的脂质代谢和炎症参与DPN 的发病机制:施万细胞线粒体功能障碍使得脂肪酸合成转向脂质氧化,进而促使髓鞘磷脂早期消耗和酰基肉碱脂质中间体的积累,导致轴突变性和神经病变[8]。此外,人源性施万细胞暴露于高糖环境后降低磷脂的合成,可被醛糖还原酶抑制剂(aldose reductase inhibitors, ARI)改善[3],暗示高糖促进了施万细胞脂质代谢失调。
施万细胞是免疫活性细胞,因其表达多种Toll样受体(Toll-like receptors, TLR)和RAGE 并产生参与DPN 发生的细胞因子,糖尿病病人血浆中被糖化修饰的脂蛋白可与施万细胞的TLR 和RAGE 结合并驱动炎症反应。在损伤后,表达多种细胞因子和趋化因子,有助于施万细胞募集免疫细胞并促进糖尿病中的神经炎症[3]。一项研究表明,葡萄糖刺激施万细胞产生的趋化因子CXCL-9,CXCL-10 和CXCL-11 可诱导T 细胞向糖尿病神经病变病人的神经聚集[9],从而促进神经病变的进展。由于炎症细胞因子可以使Aδ 和C-纤维敏感,因此这些结果也佐证了施万细胞可能参与疼痛性DPN 的发展。
2.结节区域
轴突和施万细胞之间相互作用调节髓鞘和结节域的形成。这些结节域分为三个不同的区域:Ranvier, paranodes 和juxtaparanodes 结节。结节区域富含胰岛素受体,葡萄糖转运蛋白,Na+和K+通道以及线粒体,这些均与DPN 的发展有关[2]。有髓周围神经的结节区域代谢旺盛,且是施万细胞和轴突之间相互作用最强的区域,因此易受到高糖的影响[4],且该区域富集的Na+和K+通道参与PDPN。
(1)结节区域的胰岛素受体:胰岛素与其他生长因子一起在受损轴突的神经营养支持和神经元再生中起着至关重要的作用,两者都受到糖尿病的影响[2]。最近的数据显示,长期胰岛素刺激会导致大鼠脊髓背根神经节(dorsal root ganglion, DRG)的神经元对急性胰岛素刺激反应下降(Kim B 等,2011)。这表明神经元可能产生了胰岛素抵抗。神经元对胰岛素的神经营养特性的反应性降低,导致损伤后再生潜能受损[2],这可能与结节区域的胰岛素受体有关。
(2)结节区域的葡萄糖转运蛋白:PNS 中神经元和施万细胞通过细胞膜的葡萄糖转运蛋白(glucose transporter,GLUT 家族成员转运葡萄糖,神经元中的主要类型是GLUT3[2],且GLUT3 主要在结节区域富集。糖酵解产生的乳酸是神经系统中的关键能量来源,由单羧酸转运蛋白(monocarboxylate transporters, MCT)转运,然而,糖尿病病症可影响GLUT,MCT 的定位[2]。例如,研究表明在小鼠小脑神经元中存在受胰岛素调节的易位囊泡区,类似于外周胰岛素敏感组织的GLUT4 储存囊泡;当受到胰岛素刺激时,囊泡内的GLUT 可易位至细胞表面摄取葡萄糖,为神经元提供能量(Bakirtzi K 等,2009)。然而高胰岛素血症时,神经元可出现胰岛素抵抗,导致GLUT 无法易位至细胞表面,从而影响神经元和施万细胞的能量代谢。
3.内质网应激
内质网(endoplasmic reticulum, ER)是蛋白质包装和脂质生物合成所必需的,并且充当细胞内钙储存和细胞应激的传感器。一些应激物如氧化还原状态改变,营养缺乏,葡萄糖升高和钙稳态扰动导致ER 腔内未折叠或错误折叠的蛋白质积累,导致ER应激和未折叠蛋白反应(unfolded protein response,UPR)的激活[10,11]。
在Zucker 糖尿病肥胖(Zucker diabetic fatty, ZDF)大鼠模型和链脲佐菌素(Streptozocin, STZ)诱导的糖尿病大鼠模型中,ER 应激的标志物:葡萄糖调节蛋白BiP/GRP78 和未折叠蛋白反应的GRP94 在坐骨神经中升高[10]。在三甲胺氧化物(可减轻ER应激的一种化合物)治疗的Zucker 大鼠中,在没有改善葡萄糖耐量的情况下外周神经功能障碍缓解,表明周围神经系统中的ER 应激促进了DPN[11]。对高脂饮食喂养的小鼠施用Salubrinal(可抑制ER 应激)可使其葡萄糖耐量改善,血清甘油三酯浓度恢复正常,高胆固醇血症和周围神经功能障碍减轻,坐骨神经的运动神经传导速度和后肢感觉神经传导速度改善,触诱发痛缓解[10]。这些研究均提示ER应激参与DPN 和PDPN 的病理过程。
此外,ER 参与周围神经的氧化应激。过量的UPR 活化会降低还原型谷胱甘肽:氧化性谷胱甘肽比例,降低其抗氧化能力[11]。UPR 介导的C/EBP同源蛋白(C/EBP homologous protein, CHOP)升高也消耗谷胱甘肽抗氧化能力并增加活性氧的产生,进一步提高细胞氧化应激水平。在大鼠STZ 诱导的糖尿病后12 周内用三甲胺氧化物伴侣蛋白处理降低ER 应激和CHOP 表达,减弱了坐骨神经中的脂质和蛋白质氧化,同时改善了糖尿病动物的神经病变[12]。这些研究表明了ER 在DPN 中的关键作用。
二、PDPN 的相关机制
PDPN 以自发性疼痛、痛觉过敏、痛觉超敏和一定程度感觉缺失为特征,性质为典型的神经病理性疼痛,强度异常剧烈,对标准化镇痛治疗效果差,是疼痛临床控制的重要难题。临床研究表明,在所有1 型或2 型糖尿病病人中,多达60%的病人出现神经病变,这些病人中> 30%发生神经病理性疼痛[13]。DPN 主要是感觉神经障碍,只有在疾病的后期阶段才有运动神经功能障碍。为什么感觉轴突比运动轴突更容易受糖尿病的影响?下面将讨论PDPN 的潜在机制。
1.周围感觉神经的易感性
(1)位置的差异:感觉神经元位于血脑屏障之外的脊髓背根神经节内,而运动神经元位于受血脑屏障保护的脊髓腹侧角。
(2)无髓纤维和有髓纤维的结构在糖尿病早期即发生改变。无髓C 纤维最早发生改变,且在很大程度上更容易受到代谢损伤,因为它们缺乏施万细胞的保护和营养支持[4]。 C 纤维的退化和再生导致异常性疼痛和感觉过敏。随着时间的推移,退化超过了再生,并出现C 纤维的损失,这是糖尿病病人常见的早期病变。随着小纤维病理学的进展,开始出现轻度节段性轴突脱髓鞘和髓鞘再生,进而引起神经病理性疼痛。这种神经病理学的发展过程佐证了施万细胞对轴突的保护和营养支持作用的丧失会导致最终的感觉神经轴突变性。
2.离子通道的表达致神经元兴奋性升高
钠通道在动作电位的产生和传导中起关键作用,且是大多数可兴奋细胞的电信号传导的决定因素。目前,研究证实钠通道(Nav1.1-Nav1.9)中的Nav1.7、Nav1.8、Nav1.9 参与整个伤害感受途径中动作电位的产生和传导,可引起痛性神经病变。在实验性PDPN 的早期阶段,DRG 神经元中电压门控通道的表达改变,异常电流和伤害性感受器放电增加,从而使周围神经的兴奋性增加[14]。高血糖可通过降低Na+/ K+-ATP 酶活性直接诱导周围神经过度兴奋,改变Kv1.2 钾通道亚基的分布和功能,并增加钠通道电流,这些变化共同导致伤害感受器的过度兴奋,是痛性糖尿病神经病变的关键机制[14]。此外,最近发现表明PDPN 的Nav1.8 表达增加,由此介导的TTX 电流的增加可减少C 纤维伤害感受器的传导缺失,增强对CNS 的冲动传导并促成神经病理性疼痛的发生(Sun W 等,2012)。
甲基乙二醛是一种晚期糖基化终产物,在PDPN 的病人血清中增加。当该化合物从人糖尿病血清中分离并注射到糖尿病小鼠中时,会引起小鼠出现热和机械性痛觉过敏[15],这可能是通过对钠通道和TPRA1 受体的修饰实现的。甲基乙二醛与Nav1.8通道的精氨酸残基结合, 导致Nav1.8 的失活减少,使伤害感受器过度兴奋;与Nav1.7 通道结合,促使其失活,进而增加无髓C-纤维的兴奋性[2,4,14]。TRPA1,瞬时受体电位阳离子通道亚家族A 成员1,是一种配体门控离子通道,与疼痛性DPN 的发病机理有关[4]。与TRPA1 的许多激动剂一样,甲基乙二醛可以改变TRPA1 中的关键细胞内半胱氨酸残基,导致这种离子通道的强烈激活,进而导致伤害性传入神经过度兴奋[4]。
T 型Ca2+通道调节伤害感受器的亚阈值兴奋性,且这种电流在实验性DPN 大鼠的伤害感受器中增加。在STZ 诱导的糖尿病大鼠或ob/ob 2 型糖尿病小鼠中使用特异性受体阻断Cav3.2 通道功能可以抑制痛觉过敏[16],Cav3.2 T 型通道的下调可以改善STZ 大鼠痛觉超敏反应[4]。在STZ 诱导的糖尿病大鼠模型中,神经元和神经胶质细胞均经历代谢应激和线粒体功能障碍,其导致Ca2+稳态失调,线粒体内的Ca2+缓冲受损,ER 应激导致的 Ca2+积聚和Ca2+信号传导失调,进而促进PDPN 的发展[16]。
3.小胶质细胞在PDPN 中的作用
除了周围的炎症,高血糖症和由此产生的活性氧物质也会影响脊髓的局部微环境,进而使小胶质细胞活化;反过来,激活的小胶质细胞合成并释放促炎细胞因子和神经活性分子,诱导脊髓伤害性感受神经元过度活跃,促进PDPN[17]。
(1)高血糖诱导小胶质细胞激活:高糖可激活小胶质细胞,体外研究表明,与低糖相比,高糖通过活性氧物质、PKC 和核因子κB (NF-κB)信号传导途径增加原代小胶质细胞培养基中IL-8 的分泌,提示小胶质细胞被激活[17],并且这种激活可持续6个月以上。小胶质细胞激活伴随着细胞因子的释放和丝裂原活化蛋白激酶(MAPKs)的磷酸化,包括p38-MAPK,细胞外信号调节蛋白激酶(ERK)和c-Jun N 末端激酶(JNK),它们参与痛觉过敏的产生[18]。研究表明,鞘内注射p38 抑制剂和全身大麻二酚或利多卡因可抑制p38 的磷酸化,消除了脊髓中的小胶质细胞活化以及STZ 动物的痛觉超敏反应[17]。因此,p38 磷酸化在DPN 下的小胶质细胞活化中起重要作用。
(2)激活的小胶质细胞参与PDPN:小胶质细胞激活后释放各种神经调节剂和神经活性物质,如活性氧,一氧化氮,过氧亚硝酸盐,前列腺素和促炎细胞因子,均参与DPN 的痛觉过敏和神经病理性疼痛。STZ 糖尿病大鼠的脊髓中IL-1β 和TNF-α表达增加;通过全身或脊髓给予神经胶质细胞的非选择性代谢抑制剂氟胞苷或选择性小胶质细胞抑制剂米诺环素抑制小胶质细胞,可抑制IL-1β 和TNF-α的增加以及大鼠的热和机械超敏反应(Pabreja K 等,2011)。
由高血糖诱导的激肽B1 受体(B1R)与DPN 密切相关,与野生型STZ 糖尿病小鼠相比,用STZ处理的激肽B1R 敲除小鼠无热痛觉过敏[17]。然而,在STZ 糖尿病大鼠中,脊髓背角的B1R 与小胶质细胞共表达,且激活该受体可产生热痛觉过敏。鞘内注射小胶质细胞抑制剂米诺环素和氟柠檬酸可逆转STZ 糖尿病大鼠中B1R 的上调和DPN,以及B1R 激活导致的热痛觉过敏和机械性异常性疼痛[17]。这些研究表明脊髓小胶质细胞B1R 在PDPN 中发挥重要作用。
因此,抑制小胶质细胞活化可被视为缓解PDPN 的新方法。最近一项研究发现,ammoxetine(一种新型有效的5-HT 和NE 再摄取抑制剂)对STZ诱导的糖尿病大鼠有持续的镇痛作用并可改善其抑郁样行为[18]。其通过抑制小胶质细胞的活化和p38和JNK 的磷酸化降低炎性细胞因子的水平进而发挥镇痛作用[18]。这间接证明了小胶质细胞在PDPN 中的作用。
4.巨噬细胞在PDPN 中的作用
巨噬细胞有两种极化状态:M1 和M2 巨噬细胞,分别表达大量炎症因子iNOS,IL-1β,TNF-α等和抗炎细胞因子Arg-1 等[19]。高糖状态下,M1巨噬细胞等免疫细胞被激活而表达大量炎性因子,继而导致施万细胞凋亡和PDPN 的发生。
有研究显示,抑制TNF-α 等炎性细胞因子的释放,促使M1 型巨噬细胞向M2 型巨噬细胞转化,可使链霉素诱导的糖尿病大鼠的神经传导速度,神经血流和轴突形态逐渐恢复[20]。此外,一项临床研究显示巨噬细胞大量表达iNOS 和TNF-α 的DPN病人患疼痛的风险较高,且iNOS 和TNF-α 免疫反应性越强,疼痛越严重。这些研究表明了巨噬细胞在PDPN 及神经病理性疼痛中的作用(Purwata TE等,2011)。
三、结论
DPN 研究已经从针对特定失调途径的研究(如多元醇途径通量增加,PKC 途径, AGEs 增加,血管损伤和氧化应激)发展到DPN 研究的新时代,即高血糖和由此产生的氧化应激,脂质代谢紊乱等导致施万细胞病变,损害胶质-轴突通讯和神经稳态,最终导致纤维丢失,神经变性和疼痛。轴突和施万细胞之间的相互作用在周围神经的结节域最为突出,该区域的IR,GLUT,Nav 和Kv 通道以及线粒体等多种物质均参与DPN 的形成和发展。尽管DPN 中的施万细胞病变与轴突病变之间的相互作用仍有待阐明,但目前的研究可以为DPN 的新型致病途径提供思路。ER 应激与DPN 的关联在动物模型中也获得了新的进展,ER 应激会促进周围神经的氧化应激和钙稳态紊乱,促进DPN 和PDPN。感觉神经元轴突对糖尿病损伤的易感性,钠离子通道,小胶质细胞,巨噬细胞对PDPN 的贡献也有了新的进展,但它们与DPN 发病的相关通路有何联系仍有待阐明。
猜你喜欢
浙江临床医学(2021年12期)2021-11-29 14:43:18
交通医学(2021年3期)2021-06-30 17:14:00
解剖学杂志(2021年5期)2021-03-27 12:01:41
中成药(2018年6期)2018-07-11 03:01:04
中成药(2017年8期)2017-11-22 03:18:21
吉林大学学报(医学版)(2015年1期)2015-12-17 07:47:27
长江蔬菜(2015年3期)2015-03-11 15:10:29
中华神经创伤外科电子杂志(2015年1期)2015-01-21 09:09:24
中国药理学通报(2014年2期)2014-05-09 08:22:26
西南医科大学学报(2014年6期)2014-03-20 15:43:53
