
Linking stemness with colorectal cancer initiation,progression,and therapy
2019-09-19 07:52DeepakNarayananIyerWaiYanSinLuiNg
World Journal of Stem Cells 2019年8期
Deepak Narayanan Iyer,Wai-Yan Sin,Lui Ng
Deepak Narayanan lyer,Wai-Yan Sin,Lui Ng,Department of Surgery,Li Ka Shing Faculty of Medicine,The University of Hong Kong,Hong Kong,China
Abstract
Key words:Cancer stem cell; Colorectal cancer; Tumor microenvironment; Metastasis;Extracellular matrix; Tumor heterogeneity; Resistance; Stemness; Quiescence;Recurrence
INTRODUCTION
Colorectal cancer (CRC) is a heterogeneous disease.Approximately three decades ago,the genetic roadmap for the origin and development of CRC was identified[1].Since then,massive technological progress has allowed the identification of numerous genetic and epigenetic components of the disease,further improving our understanding of the heterogeneity associated with CRC.We now know that not only does CRC exhibit a highly complex inter-tumor heterogeneity across multiple cases,warranting the need for personalized medicine[2],but it also displays a component of intra-tumor heterogeneity (morphological,genotypic,and phenotypic differences within the same tumor)[3].The overall picture was further complicated by the discovery of cancer stem cells (CSCs) that led to an altered component of intra-tumor heterogeneity (i.e.heterogeneity between clonal populations),owing to the highly dynamic nature of CSCs.Major components affecting this behavior of CSCs include tumor genetics,epigenetic signals,and most importantly,the surrounding tumor microenvironment[4].Notably,these factors have brought a landmark change in our understanding of the landscape of CRC development and progression.Considering these developments,here we review the current understanding as well as the evolving concepts of CSCs in the context of origin,development,and outcome of CRC.At the outset,we wish to clarify that the understanding of several aspects of CSCs,particularly within the field of CRC,is still at its infancy.Regardless,we aim to provide critical shreds of evidence from clinically relevant discoveries that would attempt to bridge,if not all,certain knowledge gaps existent within this field.
THE STEM CELL NICHE:PERSPECTIVES OF THE ADULT INTESTINE
An insight into the biology of intestinal stem cells (ISCs) will fuel our existing knowledge of the regulatory mechanisms of development and function of colorectal CSCs,owing to the similarities in several signaling pathways within normal and cancerous stem cells[5,6].Structurally,the intestinal epithelium is organized into several finger-like villi protrusions extending into the gut lumen that is surrounded at the base by multiple glandular invaginations,the crypts of Lieberkühn,that extend into the extracellular matrix.The villus architecture comprises of non-dividing differentiated polyclonal cells with divergent functions of nutrient absorptionenterocytes,protective mucus barrier secretion - goblet cells,and gastrointestinal hormone secretion-enteroendocrine cells; all of which,including the post-mitotic Paneth cells that reside at the bottom of the crypt,are generated from the undifferentiated,rapidly proliferating multipotent stem cells residing as monoclonal compartments within the crypts.Unequivocally,the exorbitant rate of proliferation exhibited by the ISCs within the crypts is responsible for providing a high rate of selfrenewal to the intestinal epithelium; essential to protect it by the persistent fusillade from physical,chemical,and/or biological insult[7].
Two functionally unique ISC populations are characterized within human and mice small intestine,the quiescent DNA label-retaining ISC (LRCs) identified at the +4 crypt position (characterized by the high expression of the polycomb complex protein Bmi1[8],homeodomain-containing protein Hopx[9],Tert[10],and Lrig1[11,12]markers),and the leucine-rich repeat-containing G protein-coupled receptor 5 (Lgr5+) expressing crypt base columnar cells (CBCs)[13]; both of which exhibit a self-renewal ability (selfrenewal) as well as the potency to differentiate into cells of the intestinal epithelium(multi-potency),certifying them as true stem cells[7,14].The identification of these ISC markers has been hugely possible through lineage tracing studies employing mouse models.Evidence from clonal analysis and knock-in experiments suggests that the Lgr5+CBC cells represent the actively cycling stem cell niche that gives rise to daughter cells that are transferred into a transit amplifying (TA) compartment;subsequently dividing and moving towards the crypt-villus axis as differentiated cells,promoting intestinal homeostasis[13].Contrastingly,the Lgr5-ISCs located on the+4 position of crypts are under 2% proliferative,and consequently serve as a reserve ISC pool that can assist the LRCs,directly or by regenerating the Lgr5+cell population,in case of injury from chemotherapy or radiation to the functional stem cell population[8,15].Indeed,through carefully derived genetic models,it has been shown that selective killing of Lgr5 cells can be restored by Lgr5-/Bmi1+cells located anywhere in the crypt[16].
Muñozet al[17]identified,however,that the Lgr5+cells can also express the +4 markers; suggesting a high degree of plasticity between the two ISC populations.Furthermore,secretory precursors [including the Paneth cells,enteroendocrine cells,goblet cells,and Tuft cells (IL-25 secreting chemosensory cells that increase in numbers as a type-2 immune response)] derived from Lgr5+cells expressing the Notch ligand DLL1,have also been shown to generate short-lived clones composed of differentiated secretory cells of intestinal epithelium,upon radiation-induced damage[18].Yet another regenerative mechanism was identified by Barrigaet al[19],who observed through single-cell transcriptomics,the existence of a subpopulation of Lgr5+cells expressing an RNA-binding protein Mex3a that divides at a relatively slower rate,making it more resistant to radiation or chemotherapy-induced damage,and can help regenerate all intestinal lineages.To summarize,the small intestinal crypt functions as a well-defined network of interdependent cellular niches that serve as multiple layers of backups to ensure the smooth and continual functioning of the stem cell machinery to maintain intestinal homeostasis.
Within the ISC research community,while the small intestine has received a major focus,the colon stem cells lack significant characterization[5].As compared to the small intestine,the colon shows large differences with the overall anatomy of the intestinal epithelium as well as the cellular architecture.The large intestine is devoid of any finger-like villi protrusions,or crypt based Bmi1+cells,and Paneth cells[5].The colonic stem cell niche has been characterized primarily with cells showing a high expression of Lgr5 as well as ephrin type-B receptor 2,olfactomedin-4,and achaete-scute complex homolog-2 markers; which are capable of self-renewal and giving rise to the cells of the intestinal lineage[13,20-22].Studies also indicate,however,the presence of slow-cycling colonic Lrig1+cells that attempt to replenish the Lgr5 cell population upon injury[11].Reports also show the presence of doublecortin-like kinase 1 cells within the colonic crypts that have been found to be actively proliferating in the presence of growth factors and give rise to intestinal lineage cells,forming enteroids[23,24].Furthermore,unlike small intestine where the Paneth cells serve as the primary source of wingless/integrated (Wnt) signaling molecules that guide the renewal of the epithelium,recent work by Degirmenciet al[25]showed the existence of a group of subepithelial mesenchymal cells expressing zinc finger protein Gli1 that act as a critical source for Wnt secretion that directs colonic stem cell renewal.Moreover,the crypt structural elements,specifically the colonocytes,have also been found to yield a protective function to the proliferating cells within the crypt from potent metabolites produced by the intestinal microbiota[26].Notwithstanding these crucial bits of research,a lot must be done to understand better the colonic crypt and the associated stem cell niche.
ROLE OF INTESTINAL STEMNESS IN THE ORIGIN OF CRC
The original model for colorectal carcinogenesis and progression,the “Vogelgram”,was laid down rather elaborately by Fearon and Vogelstein[1].It provided a schematic in which loss of the adenomatous polyposis coli (APC) tumor suppressor gene would result in an adenoma and subsequently mutations inKRAS,TP53,phosphoinositide 3-kinase (PI3K),and other genes would cause the development of a metastatic disease.A principal feature of CRC identified through this model was the monoclonal origin of the disease (i.e.CRC originates from the clonal expansion of one hyperproliferating cell).Importantly,the involvement of the crypt and the corresponding ISCs residing within them as CRC initiators were debated upon,since the analysis of several spontaneous adenomas found dysplastic cells with mutations inAPConly on the luminal surface of the colon,while the underneath crypt and the ISCs were normal[27].This finding caused the development of the ‘top-down' model of tumor initiation that begins at the top of the crypt,in the intra-cryptal zones between crypt orifices,and then spreads laterally and downward,displacing the normal epithelium of crypts[27].Though this was true for patients with familial adenomatous polyposis (FAP),immunohistochemical studies of early sporadic colorectal adenomas have shown proliferative adenomatous epithelium with nuclear beta-catenin within the entire crypt; pointing at the role of crypt based stem cells as progenitors of CRC[28].In parallel,the Wright lab also examined the mucosa from FAP patients post-surgery and found that both sporadic and FAP adenomas originate as a uni-cryptal adenoma,with dysplastic lesions in a single but entire crypt,and grows ‘bottom-up' by a division of the crypt at the base,termed as crypt fission.Although the lateral and downward spreading model was not completely excluded,several studies pointed out that crypt fission is the primary mode of adenoma progression in FAP[29,30]as well as sporadic adenomas[31].Indeed,an alternative explanation for the top-down model was suggested by Shihet al[27]wherein the stem cells at the base of a single crypt develop the neoplasm,which subsequently transforms and migrates up the crypt and become a part of the superficial mucosae,which then spreads laterally and downward into adjacent crypts.
It has been shown in the past that the severity of intestinal cancer depends largely on the initiation than the progression,indicating the importance associated with the early events of CRC development[32,33].Importantly,the identification of ISC specific expression markers has allowed for functional techniques that can be used to determine if they can function as progenitors of colorectal carcinogenesis.In 2007,O'Brienet al[34]characterized the first tumor stem cell marker,CD133,and pointed towards a CSC model of tumor initiation driven by CD133+cells in CRC.A plethora of studies has indicated that specific deletion ofAPCin ISCs expressing Lgr5,LRIG1,or CD133 markers can induce rapid adenoma generation[11,35-37].Moreover,activation of the β-catenin pathway within these cells as well asBMI1+cells resulted in a similar outcome,indicating that ISCs are the primary cells of origin of CRC[8].Mutations within key signaling pathways,including Wnt,Notch,and Hedgehog pathways,can dislodge the wild-type ISCs from the control of regulatory signals,allowing them to develop precursor lesions[38,39].Most of these approaches,however,caused the generation of intestinal adenomas in mice that commonly occur in the small intestine and do not generally progress to carcinoma[40,41].In contrast,human intestinal malignancies mostly occur in the colon.Additionally,the development of human CRC is also strongly dependent on environmental factors such as inflammatory conditions[42],which are lacking in genetic mouse models[5].
Owing to multiple differences between carcinogenesis in genetically modified mice and human patients,several studies have looked at the dynamics of stem cells in response to key genetic mutations and its influence on the development of CRC to improve our grasp on the actual mechanisms of CRC origin.Mutations within the intestine were assumed to arise due to several factors,including DNA replication errors and environmental factors such as carcinogen exposure,inflammation,etc.Since the TA cells represent the most actively proliferating population within the crypt,they are more prone to mutations; although given the short life span of these cells and the mild phenotype of the mutation,mutated TA cells offer a lower risk of serving as tumor initiators[43].Indeed,it was shown that the wild-type ISC division follows the neutral drift principle to replace randomly any of the other crypt ISC populations[44,45].Although oncogenic mutations aim towards preventing this,Vermeulenet al[46]and Snippertet al[47]suggest that the mutated cells can also be stochastically replaced by wild-type ISCs.What this means is that the likelihood of an inactivating mutation in a key tumor suppressor,likeAPC,to get fixed is less than 50%,making the mutated cell highly susceptible of getting lost in the continuous process of replacement[43].Such a low probability makes CRC an extremely slow disease,postulated to take over a decade for cellular mutations to accumulate that could drive the initiation and progression of the malignancy[48].Importantly,the presence of accompanying conditions such as intestinal inflammation tends to allow the mutated cells to prevail,pointing at the importance of environmental factors in conjunction with genetic factors in playing a critical role in CRC initiation[46].Not surprisingly,while a competition exists between the normal and the mutated ISCs during the tumor initiation process,disease progression is associated with a rivalry between the CSCs,with stronger clones characterized by a larger number of accumulated mutations and resistance to environmental factors such as therapy[5].
In addition to a stem cell-based CRC origin model,a few studies have indicated a role of differentiated cells in serving as the cell of origin for the disease.Like the ISC to colorectal CSC model,most of these studies also indicate that genetic events combined with environmental factors can favor the development of CRC.A loss ofAPCin the tuft cells accompanied by microenvironmental disturbances was found to induce colonic tumors[49].Moreover,transgenic mice models have shown that intestinal epithelial cells can also dedifferentiate into tumor-initiating stem cells under the influence of enhanced Wnt and the inflammatory nuclear factor-kappa B (NFκB)signaling pathway[50].Alternatively,accumulation of mutations such asKRASG12Dthat activates inflammatory signaling,accompanied with a loss ofAPCthat results in the activation of Wnt pathway,yielded similar tumor initiation from the differentiated cells[50].Taken together,the studies indicate that while CRC can have a stem cell(primary) or a non-stem cell (secondary) origin,the contributing factors include accumulation of mutations as well as environmental factors to confer a functional advantage for the development and progression of the tumor.
INFLUENTIAL PATHWAYS REGULATING THE COLORECTAL CSCs
There exists a molecular network surrounding the complex development of CSCs associated with CRC,and they are only recently emerging.Deciphering this network will not only improve our understanding of the role of stem cells in the origin and pathogenesis of CRC,rather will provide better therapeutic avenues to deal with the malignancy.Although several signaling pathways have been implicated,notable ones that have been found to play crucial roles in the growth and functional maintenance of CSCs include the Wnt,Notch and Hedgehog,and the bone morphogenetic protein(BMP) pathways.
The canonical Wnt signaling pathway has been identified as a hallmark in the regulation of stem cells-from maintenance,proliferation,differentiation to apoptosis[51].Under normal signaling,binding of the Wnt ligand to the transmembrane receptors tends to stabilize and allows the nuclear translocation of βcatenin causing transcriptional activation of important targets includingc-Myc,Axin2,Lgr5,andASCL2that govern stem cell fate,proliferation,as well as maintenance[52-56].Specifically,within the intestine,active Wnt signaling is essential to maintain the stem cell niche within the crypt and promote gut homeostasis[57].Intuitively,abnormal Wnt signaling has been implicated in several cancers,including CRC[58].Inactivating mutations inAPCand consequently a hyperactive Wnt signaling or activating mutations in β-catenin have been found in most of the CRC cases and has been identified as one of the initiating steps in tumor development[59].In line with the ISC as the CRC cell of origin theory,Vermeulenet al[60]reported that CD133+CRC cells growing as tumor spheres in culture contain a subpopulation of cells with constitutively high Wnt signaling.However,only a subset of these cells with the highest Wnt signaling was observed to show nuclear localization of β-catenin and behaved as CSCs.Denoted as the “β-catenin paradox”[61],the existence of intratumoral heterogeneity of Wnt signaling indicated that the pathogenesis of CSCs in CRC required contribution from other factors in addition to the loss ofAPC,such asKRASmutations[62],PI3K[63],Notch[64]and Hedgehog signaling[65].Moreover,mutations in essential Wnt pathway components,including the R-spondin/Lgr5/RNF43 module have been identified in almost 1/5thof CRC cases,which commonly co-occur withAPCinactivation/deletion[66].
Importantly,Lgr5+cells have been found to propagate CSCs within colon adenoma,and subsequently,Lgr5 has been identified as an important CSC marker[67].More recently,studies have pointed out that while Lgr5-cells can revert into an Lgr5+cell phenotype,allowing the development,maintenance,and metastasis of the growing tumor[68]; inhibition of Lgr5 strongly suppressed the growth of patient-derived tumor organoids[69].These findings suggest that Lgr5+CSCs are detrimental for the growth and propagation of CRC.Furthermore,Myantet al[70]show that followingAPCloss,the small GTPase RAC1 helps in the propagation of Lgr5+CSCs in colon cancer by activating reactive oxygen species production,which activates NFκB signaling that promotes Wnt signaling.Co-activation of NFκB signaling and Wnt signaling has also been shown to promote colorectal tumorigenesis by causing dedifferentiation of intestinal cells into stem cells[50].
Cross-talk has also been observed between the Wnt signaling pathway and critical members of the Notch pathway.Like Wnt,Notch signaling is predominantly higher within the stem cell populations of the crypt and gradually decreases in the differentiated compartment,suggesting that Notch also contributes to ISC maintenance.An early study by the Clevers group[71]on ApcMinmice carrying a heterozygous mutation forAPCthat causes multiple intestinal neoplasia,identified a collaboration between active Notch signaling and Wnt pathway that is indispensable to maintain the proliferative adenoma cells.Moreover,suppression of Notch signals by deletion or by inhibition with a γ-secretase inhibitor resulted in an increase in the levels Math1,a basic helix-loop-helix transcriptional activator of cell differentiation in the intestine[72],consequently causing the arrest of cell proliferation within the crypt and the conversion of the crypt cells into differentiated secretory goblet cells[71].It has been indicated,however,that goblet cells are commonly absent in CRC and show downregulated expression of Hath1,the human orthologue of Math1,suggesting an active Notch signaling in most CRC cases[73].
In contrast to the Wnt and the Notch pathways,the Hedgehog and BMP pathways are primarily active within the differentiated cells of the crypt.Although Hedgehog genes are commonly upregulated in CRC[74,75],numerous studies indicate that Glidependent canonical Hedgehog pathway antagonizes Wnt signaling promoting tumor cell differentiation[65,76,77].This makes it difficult to treat CRC with drugs targeting members of the Hedgehog signaling since this strategy seemed to promote Wnt-based proliferation of CSCs[78,79].However,recently,Reganet al[80]clarified that while the Gli-dependent Hedgehog signaling downregulates Wnt signaling,the noncanonical PTCH1-dependent Hedgehog signaling promotes Wnt signaling to allow the maintenance of CSCs in CRC.This breakthrough lets physicians target the two pathways (Hedgehog and Wnt) independently,allowing improved management of the disease.
Like Hedgehog,the transforming growth factor (TGF)-β/BMP pathway has been found to have diverse associations with the Wnt signaling network:From inhibition -by promoting cell differentiation and apoptosis[81],and to collaboration - by causing CRC tumorigenesis[82].More recently,it was demonstrated that BMP signaling inhibits Lgr5 stem cell signature through a Wnt signaling independent mechanism by SMAD1/SMAD4 recruitment of histone deacetylase that blocks transcription of key factors essential to maintain the stemness of CSCs[83].Indeed,germline mutations within the BMP receptor type I or its downstream effector SMAD4 have been shown to have a high risk of CRC[84,85].Additionally,Whissellet al[86]reported a key role for the zinc-finger transcription factor GATA6,which was found to help maintain the Lgr5+CSCs in adenoma,simultaneously suppressing BMP signaling by blocking the binding of β-catenin/TCF4 transcriptional complex to a regulatory region of the BMP4 locus within the differentiated tumor cells.In vivoknockdown of GATA6 was found to upregulate BMP signaling,suppressing CRC development.
Put together,these pathways offer a telescopic view of the multiple mechanisms of regulation of stem cells in the origin and development of CRC.Only by improving our understanding of these mechanisms of CSC regulation can we advance the therapeutic strategies required to deal with the progress of the disease.
ROLE OF STEMNESS IN CRC METASTASIS
Since stem cells have been implicated as the primary cell of origin of CRC,it is safe to assume that CSCs originating from ISCs would play a crucial role in the maintenance as well as the spread of the disease to distant sites.In 2010,our laboratory published the earliest account of a subpopulation of CD26+CSCs from a primary CRC tumor responsible for the development of distant metastasis[87].An important observation was the discovery of the ability of CD26+CSCs isolated from the CRC tumor of a patient with liver metastasis to cause the formation of liver metastasis in mice,regardless of their CD133 or CD44 expression status.High expression of CD26 was also found to be associated with advanced tumor staging and poor overall survival of the patients[88].While CSCs were considered as the key factors responsible for branching and spreading of the primary tumor,it was interesting to note that only small sub-populations of CSCs could initiate metastasis.Indeed,Brabletzet al[89]had deduced the existence of two subgroups of colorectal CSCs:The stationary CSCs that remain active within the primary tumor yet cannot disseminate to newer sites and,The mobile CSCs that are derivatives of stationary CSCs,but can form metastatic colonies.Importantly,for a CSC to be considered within the second group,the CSCs must have undergone epithelial-mesenchymal transition (EMT) to disseminate and form metastases while retaining its self-renewal capacity,heterogeneity acquired from the asymmetric division,as well as plasticity to adapt to the newer environment[89,90].Moreover,a higher number of mobile CSCs at the tumor-host interface associated with the EMT phenotype has been found to correlate with an overall poor disease prognosis[91,92].
While distinct populations of CSCs initiated tumor progression,it was also essential to identify whether the same group of cells colonized target organs at random or through a tight-knit molecular pathway.Since the colorectal CSCs commonly enter the mesenteric circulation,metastasis is more often observed in the liver,followed by the lungs[93].An elegant study by Gao and colleagues characterized CRC-specific migrating CSCs responsible for organ-specific metastasis[94].The authors identified a specific group of CSCs expressing CD110,the thrombopoietin receptor that caused liver metastasis; considering that liver is the primary site for thrombopoietin production and hence serves as a chemotactic signal for the CD110+CSCs.Furthermore,CSCs expressing CUB-domain-containing protein 1 could alone colonize the lungs by homing to the lung endothelial cells[94].Thus,metastatic colonization not just depends on the existence of specific markers on the initiator cells but requires a specific complement of the target biological organ with prometastatic functions.
Although CSCs were hypothesized as the primary force for CRC progression and metastasis,it was not clear how this trait was orchestrated.What are the genetic or epigenetic or environmental mechanisms forcing the conversion of CSCs from tumor instigators to propagators? A crucial step towards malignancy is the induction of the EMT pathway-A fundamental process of embryonic development as well as cancer metastasis,characterized by the loss of the epithelial morphology and the accompanying markers simultaneously acquiring the mesenchymal phenotype.The process is primarily driven by the activation of a cohort of transcription factors,including snail,zinc-finger E-box binding factor,twist,and several others[95].Accumulation of key genetic mutations and epigenetic changes in combination with an invasive environmental signal triggers the formation of migratory CSCs that show a high expression of genes critical for EMT as well as for maintaining the CSC phenotype,such as Slug,β-catenin,N-cadherin,as well as Lgr5,CD133,and CD44[90].Importantly,Brabletzet al[89]suggested that microenvironmental alterations have a greater say over genetic factors in inducing EMT,since a reduction of these signals at the target site reverses the EMT pathway,allowing organ colonization.Indeed,the presence of pro-CSC microenvironmental cytokines,including,stromal cell-derived factor 1,osteopontin,and hepatocyte growth factor,promote the activation of PI3K and nuclear accumulation of β-catenin,causing a concomitant increase in the migratory metastatic CSC pool[96,97].In fact,the pro-CSC cytokines are known to induce CSC plasticity by causing the dedifferentiation of tumor cells into a CSC phenotype that may subsequently adopt the metastatic CSC feature[60,97].
Another study identified the influence of tumor-associated macrophages secreted milk-fat globule-epidermal growth factor-VIII in conferring the CSC with a selfrenewal and chemoresistance ability,by activating the Stat3 and Sonic Hedgehog pathways in CSC populations[98].Contrarily,increase in the levels of the BMPs,a tumor suppressive cytokine,promotes CSC differentiation,inhibits the Wnt pathway by upregulating the phosphatase and tensin homolog and suppressing PI3K[99],and limits the expression of CD44v6; a diagnostic marker of metastatic CSCs[97].Interestingly,the BMPs belong to the TGF-β superfamily and tend to inhibit tumor progression,while Todaroet al[97]suggest that TGF-β may contribute to the metastatic activity of CD44v6+ cells.Indeed,TGF-β serves as a key microenvironmental factor that plays dual roles,serving as a tumor suppressor during the early transformation phase and subsequently playing a pivotal role as an oncogene during the progression phase; a switch catalyzed by the accumulation of key mutations,such as p53[100]and SMAD4[101].
An early study by Calonet al[102]showed that the migratory CSCs ready for colonization can instruct the stroma of the host organ,by inducing an increase in the levels of TGF-β eitherviaactive secretion or,indirectly by the recruitment of macrophages,cancer-associated fibroblasts or platelets that produce TGF-β.Moreover,by activating the Smad proteins,specifically Smad2,Smad3,and Smad1/5/8,TGFβ1 has been shown to induce both EMT as well as stemness in CRC cells,leading to liver metastasis[103].Additionally,a functional loss of Smad 4,a critical member of the TGF-β/Smad signaling,has been found to correlate with an increase in EMT signaling characterized by the loss of E-cadherin,leading to distant metastasis and overall poor patient prognosis[104-106].Oncogenic TGF-β has been found to cooperate with a hyperactive Raf/mitogen-activated protein kinase pathway to cause an EMT phenotype[107].Moreover,a collaboration between the loss of the epithelial Ecadherin protein and an increase in Wnt/β-catenin signaling,with an increased TGFβ release,allows cells to maintain the mesenchymal phenotype in the EMT process[108].Following dissemination of the CSCs to the target organs,the new microenvironment is generally hostile towards the incoming tumor cells,which may affect the stemness as well as the survival of the cancer cells.CSCs in CRC have been reported to suffer apoptosis almost immediately after reaching the liver[102].Survival environmental signals in the form of cytokines and growth factors are hence generated by infiltrating cells from the primary tumor,along with the activation of stemness promoting Wnt as well as Notch pathways,to promote the creation of pre-metastatic niches within the target organs and improve the endurance of CRC based CSCs as well as allow colonization.A model of the role of stem cells in the normal colon,CRC development,as well as metastasis is shown in Figure 1.
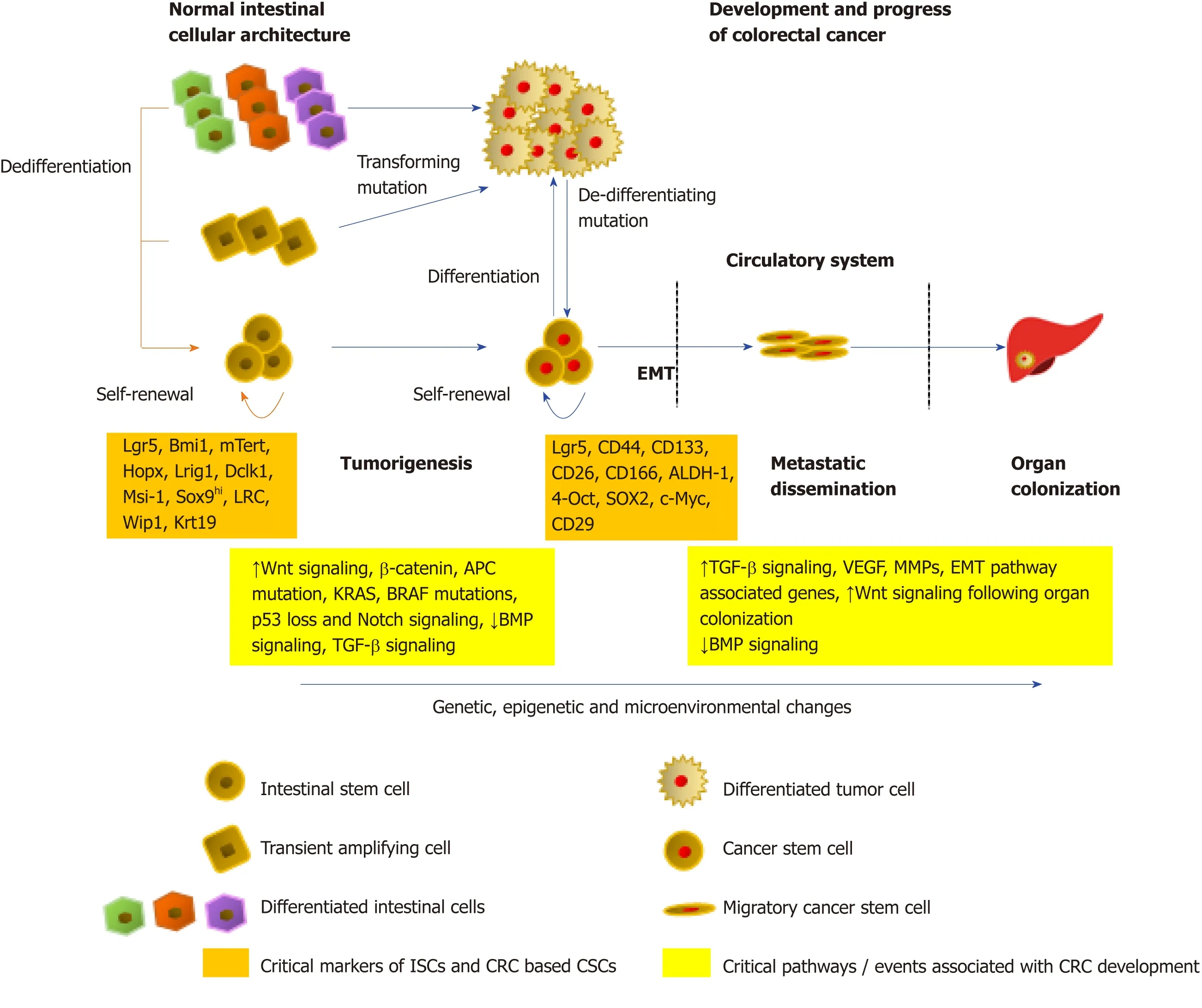
Figure1 Role of stem cells in normal colon and CRC.
THE RELEVANCE OF STEMNESS IN CRC PROGNOSIS AND THERAPY
Several factors affect the outcome in CRC—cancer spread (metastases),chemoresistance,and recurrence; all of these being mutually exclusive events.Moreover,numerous studies have pointed out a stronger involvement of CSCs,accompanied with our current inefficiency in understanding the biology of these cells in the malignancy,in allowing the factors to become dominant,leading to poor disease prognosis.Traditional therapy tends to debulk the tumor off the mature,differentiated cells,while the CSCs stay quiescent,and hence become resistant to drug or radio-therapy,allowing for improved opportunities to promote recurrence.An early report in this direction identified that the quiescence of CSCs can be attributed to an increased expression of ATP-binding cassette drug transporters,active DNArepair machinery,as well as an innate resistance towards apoptotic cell death[109].Furthermore,variants of key CSC associated markers including Lgr5,CD44,and aldehyde dehydrogenase 1A1 have been found to be associated with a shorter time to tumor recurrence in high-risk stage II and stage III CRC patients treated with fluoropyrimidine-based therapy; suggesting the association of the variants with improved survival and chemoresistance abilities of the CSCs[110].It is important to realize that while traditional adjuvant fluoropyrimidine and/or platinum-based therapy has been found to be effective in CRC,exposure to these chemical agents may enrich a pool of CSCs responsible for resistance and recurrence.As proof of principle,the treatment of patients with unresectable CRC with mFOLFOX6 therapy was found to increase the levels of several CSC markers in distant metastases[111].A previous study in this direction made use of cyclophosphamide or Irinotecan-based chemotherapy to treat xenogeneic CRC tumors and subsequently found an enrichment of a group of drug-resistant CSC populations with elevated levels of ALDH1 that could regenerate tumors[112].
While more aggressive CSCs tend to emerge in tumor development,it is the feature of plasticity that allows CSCs to pioneer resistance to therapy as well as recurrence.A study by Kobayashiet al[113]identified the interconversion of CSCs from a proliferative Lgr5+state to a quiescent,drug-resistant Lgr5-state in the presence of an anticancer drug.Following reseeding and drug removal,the Lgr5-cells transitioned back to the Lgr5+state,while maintaining thein vivotumor-initiating properties all the time in both states.It is hence essential to identify and target key molecular factors that are common to multiple states of CSCs to achieve a better therapeutic cleanup in CRC.Indeed,by gene profiling studies the authors demonstrate that an epidermal growth factor receptor ligand,epiregulin,is expressed by both the Lgr5+and Lgr5-states that could be targeted using an anti-epiregulin antibody[113].Several reports have also indicated that the resistance and recurrence abilities of CSCs are strongly influenced by the tumor microenvironment as well as key signaling pathways and epigenetic modifications.Numerous cytokines and chemokines secreted by cancer-associated fibroblasts,particularly the MET receptor ligand hepatocyte growth factor,were found to promote CSC proliferation while making them resistant to apoptosis in response to epidermal growth factor receptor therapy[114].Overactivation of Wnt signaling,a critical signaling network in the growth and development of stem cells,has been observed in 5-fluorouracil resistant CRC,while downregulation of Wnt transcription factor T cell factor 4 increases the sensitivity of the tumor to radiation therapy[115].
Recently,the role of microRNAs has also been identified as potent modulators of stem cell signaling within CRC.Notable ones include miR-15a and miR-16-1,which are frequently deleted in CRC cell lines as well as clinical specimens,are found to be associated with a greater number of B cells positive for immunoglobulin A (IgA+B cells) and shorter survival periods[116].At the molecular level,deletion/inhibition of miR-15a/miR-16-1 results in the upregulation of AP4,a c-Myc target[117],through a double negative feedback loop,resulting in distant metastases and poor survival[118].MiR-15a has also been found to impact several other key genes implicated in the origin,maintenance,as well as chemoresistance of CSCs in CRC,including YAP1,doublecortin-like kinase 1,BMI1,and BCL2[119].Similarly,the expression of miR-16-1 is negatively correlated with cyclooxygenase-2 level[120]which is also a downstream effector of the Wnt signaling pathway and thus has an active role in regulating the stem cell biology in CRC.Altogether,the miR-15a/miR-16-1 complex serves as a valuable therapeutic target to specifically tackle pathways associated with CSC maintenance in CRC.
Successful CRC targeting requires the inhibition of key pathways and environmental signals that function to promote the self-renewal ability,apoptotic resistance,stemness,as well as prolonged survival of the CSCs.Several potential CSC targeting drugs have been identified in the past several years,a few of which are under trial as well.Studies by Todaroet al[121,122]demonstrated a mechanism of apoptosis evasion by CD133+CSCs by expressing IL-4,which could be neutralized by the treatment of the cells with an anti-IL4 antibody,IL-4DM.Moreover,silencing of the Aurora-A kinase,a critical regulator of mitosis,has been found to affect the colorectal CSCs by inhibiting proliferation,promoting the apoptotic potential,and sensitizing the cells to chemotherapy[123].The role of mitochondrial targeting molecules as potential therapeutic agents has also been identified.A remarkable study by Colaket al[124]identified a role of BCLXL in protecting colon CSCs from chemotherapy,determined by decreased mitochondrial priming.By making use of BH3 mimetics,the authors successfully inhibit the BCL2 family members,sensitizing the CSCs to chemotherapy.Additionally,several molecules targeting critical members of Notch signaling[125,126]as well as the Wnt pathway[127]have been identified.
Although there are many more therapeutic targets as well as potential drugs under pre-clinical/clinical trials,understanding the clinical phenotype of the patient is critical to the usage of these drugs.Recent studies focusing on the development of CSC-targeting drugs advise upon the combined use of these drugs with the conventional adjuvant therapy to maximize the potential (Figure 2).The efficiency of CSC-targeting drugs is particularly higher on circulating CSCs due to the absence of a safe microenvironment as well as the presence of a toxic adjuvant therapy[5].In advanced CRC,debulking of the tumor would not directly correspond to a similar loss of volume of the associated CSCs.In addition,in aggressive tumors,combination therapies increase the stress on tumor microenvironment,which has been known to contribute to an increase in the CSC pool.The situation is made further complex by the ability of CSCs and differentiated cells to interconvert.Strategies for monitoring the efficacy of CSC-targeting are still at infancy.Though several CSC markers have been identified in CRC,most of them are also expressed by ISCs.The success of CSCtargeting drugs hence strongly depends on the improvement of CSC monitoring techniques.Additionally,studying of patient-derived models of CRC is essential to increase our knowledge of the roles of CSCs and help piece the missing gaps within this field.
CONCLUSION
The landscape of CRC has progressed from a simple hierarchical model to a complex setup interspersed with multiple roles of dynamic CSCs that are modulated constantly by genetic,epigenetic,and specifically,microenvironmental factors.Although the discovery of CSCs in CRC was made roughly a decade ago,our understanding of the biology of these cells is still quite limited[34].With the progress of technology,our existing knowledge of the complex roles and the dynamic nature of colonic stem cells,as well as CSCs,is undergoing constant evolution.However,better techniques for detection and isolation as well as the usage of patient-derived CRC models is essential to further our understanding of CSCs in CRC.
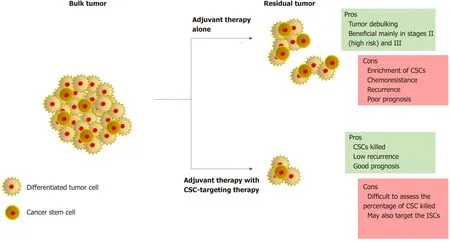
Figure2 Therapeutic strategies in colorectal cancer.
 World Journal of Stem Cells2019年8期
World Journal of Stem Cells2019年8期
- World Journal of Stem Cells的其它文章
- Moving forward on the pathway of cell-based therapies in ischemic heart disease and heart failure - time for new recommendations?
- Neural regeneration by regionally induced stem cells within poststroke brains:Novel therapy perspectives for stroke patients
- Orchestrating stem cell fate:Novel tools for regenerative medicine
- Bone marrow microenvironment:The guardian of leukemia stem cells
- Tonsil-derived stem cells as a new source of adult stem cells
- Derivation and applications of human hepatocyte-like cells