
Bone marrow microenvironment:The guardian of leukemia stem cells
2019-09-19 07:52MohammadHoushmandTeresaMorteraBlancoPaolaCircostaNarjesYazdiAlirezaKazemiGiuseppeSaglioMahinNikougoftarZarif
World Journal of Stem Cells 2019年8期
Mohammad Houshmand,Teresa Mortera Blanco,Paola Circosta,Narjes Yazdi,Alireza Kazemi,Giuseppe Saglio,Mahin Nikougoftar Zarif
Mohammad Houshmand,Paola Circosta,Giuseppe Saglio,Department of Clinical and Biological Sciences,University of Turin,Turin 10126,Italy
Teresa Mortera Blanco,Mahin Nikougoftar Zarif,Center for Hematology and Regenerative Medicine,Karolinska Institutet,Department of Medicine,Karolinska University Hospital Huddinge,Stockholm 14183,Sweden
Narjes Yazdi,Department of Molecular Genetics,Tehran Medical Branch,Islamic Azad University,Tehran 1916893813,Iran
Alireza Kazemi,Department of Hematology and Blood Banking,School of Allied Medical Sciences,Shahid Beheshti University of Medical Sciences,Tehran 1985717443,Iran
Mahin Nikougoftar Zarif,Blood Transfusion Research Center,High Institute for Research and Education in Transfusion Medicine,Tehran 146651157,Iran
Abstract
Key words:Bone marrow microenvironment; Bone marrow niche; Leukemic stem cell;Chronic myeloid leukemia; Acute myeloid leukemia; Target therapy
INTRODUCTION
Chronic myeloid leukemia (CML) is a clonal hematopoietic stem cell (HSC) disorder,emanating from t(9;22)(q34;q11.2),a translocation that involves fusion of Abelson murine leukemia viral oncogene homolog 1 (ABL1) on chromosome 9 and breakpoint cluster region protein (BCR) on chromosome 22[1].The encoded protein by constitutive tyrosine kinase activity stimulates downstream signaling pathways that lead to increased expansion of leukemic cells.Although the chronic phase of CML is concomitant with normal cell maturation,in the absence of appropriate treatment,a second mutation transforms the chronic phase into acute phase that mimics the same pattern asde novoacute leukemia[2,3].
Acute myeloid leukemia (AML) is the most common form of leukemia in adults and is characterized by perturbed proliferation,block of differentiation,and infiltration of leukemic cells into the bone marrow and blood[4].Current therapies result in overall survival of about 40% in patients younger than 60 years of age,while this rate declines in older patients to 5%-15% and is associated with higher morbidity and mortality[5].One major concern in the treatment of AML is drug resistance,and a promising approach such as targeted therapy for relapsed or refractory AML is of the essence.While in CML the introduction of tyrosine kinase inhibitors (TKIs) as a milestone in the treatment of CML results in overall survival of about 86% and attaining treatment-free remission (TFR) seems achievable[6].
Common treatment of AML and CML is based on the elimination of bulk disease population[7].As propagation of resistant leukemic cells may continue after the treatment discontinuation,the concept of cancer stem cell (CSC) came to light.Based on this theory,a cell with the self-renewal capability and leukemic related genetic alterations,which stands at the apex of the hierarchy,may be able to resist to therapy and sustain the relapse of the disease later on[8](Figure 1).The first approach that proved the existence of CSC was in AML,where the transplantation of a small cell population with stem cell-like properties into non-obese diabetic/severe combined immunodeficiency mice culminated in leukemia[9].The fact that every cell in different stages of the maturation by gaining stem cell-like features has the potential to become CSC is of paramount importance and depicts that it is not crucial for CSC to have stem cell origin[10].
While both CML and AML leukemia stem cells (LSCs) have distinctive characteristics in case of the biology and immunophenotype,they share common properties such as drug resistance,quiescence,heterogeneity,and the microenvironment they reside.The bone marrow microenvironment (BMM) underpins normal hematopoiesis by secreting various growth factors and physical interactions with HSCs and progenitor cells[11].In AML and CML,the BMM boosts leukemogenesis through an interaction with LSCs,and in turn,LSCs change the BMM based on their requirements and make it less hospitable for normal stem/progenitor cells[12].Considering BMM as the main sanctuary for LSCs,targeting these interactions may provide an ample opportunity to treat leukemia more effectively.In this review paper,we focus on the protective role of the BMM in the survival of CML and AML LSCs.We then move toward specific markers to identify these cells and put forward possible ways to target them within the BMM.
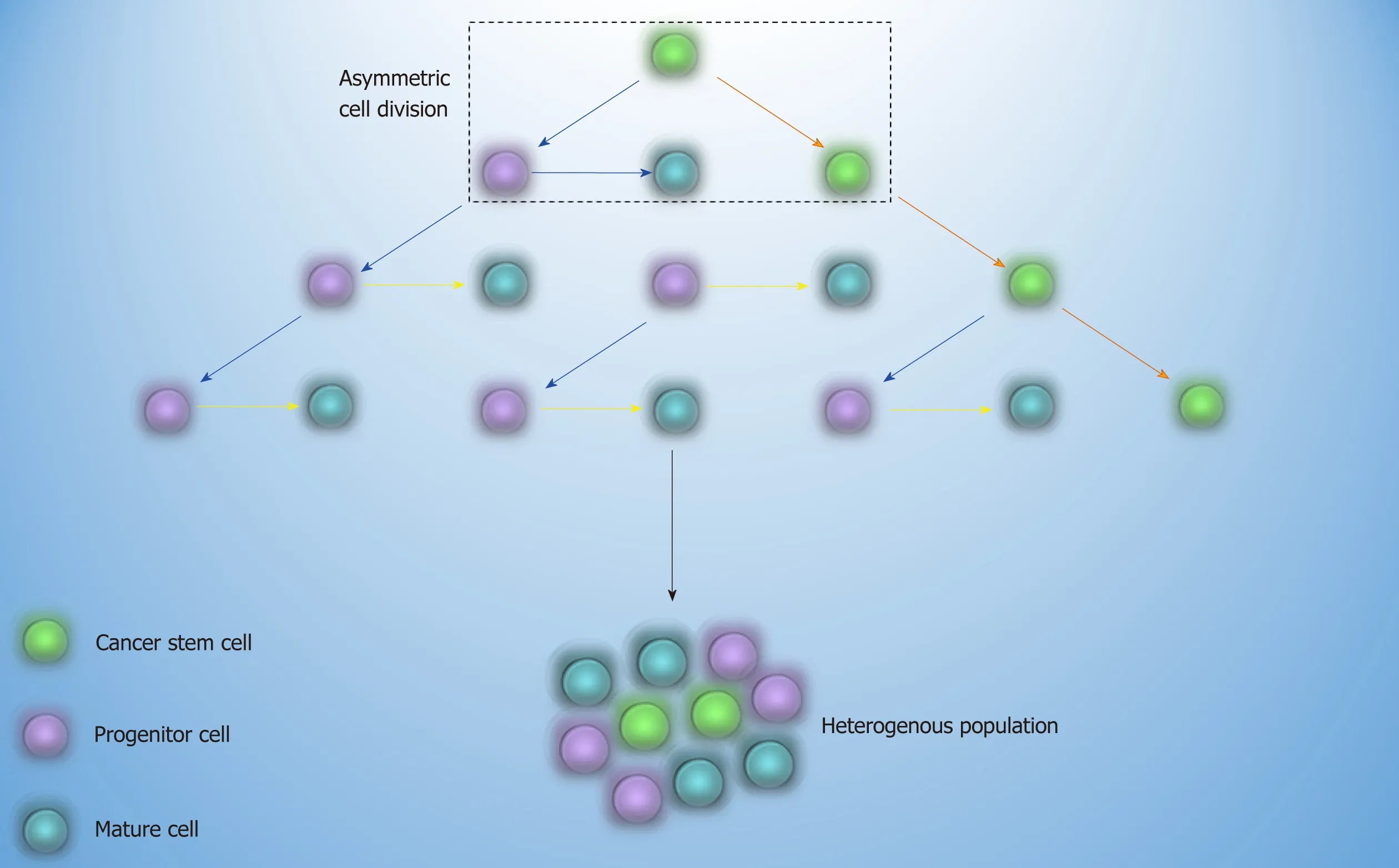
Figure1 Cancer stem cell theory.
CML LSCs AND BONE MARROW MICROENVIRONMENT
CML LSCs,due to their resemblance to normal stem cells,reside in the same microenvironment in which a reciprocal relationship between these cells and components of the BMM is linked with enhanced proliferation,quiescence,and drug resistance.All of these mechanisms are conducted by sets of adhesion molecules or secretion of cytokines,chemokines,and growth factorsviaparacrine or autocrine mechanisms.
C-X-C motif chemokine ligand 12 (CXCL12),a known chemoattractant for the homing process,is secreted by mesenchymal stromal cells and osteoblastic cells and has a role in the localization of CML LSC and normal HSC in the BMM[13].However,perturbed expression of C-X-C chemokine receptor type 4 (CXCR4) by CML LSCs or CXCL12 targeting by CML LSCs impacts the homing process.Kinase activity of P210BCRABL1and activation of downstream signaling pathways,such as phosphoinositide 3-kinases/protein kinase B [PI3K/PKB(AKT)],result in downregulation of CXCR4 by CML cells[14].Moreover,increased secretion of granulocyte-colony stimulating factor (G-CSF) as an antagonist of CXCL12 by CML LSCs[15]and aberrant expression of surface marker dipeptidyl peptidase 4 (CD26) on CML LSCs with a chemokine cleavage activity favor mobilization of CML LSCs into the blood[16].However,TKIs,by inhibiting P210BCRABL1,contribute to the upregulation of CXCR4 and migration of CML LSCs to the BMM[14].
The homing process for normal HSCs initiates with tethering and rolling of HSCs on endothelial cellsviainteraction with P and E-selectin.Then,a strong attachment through very late antigen-4 (VLA-4) and VLA-5 with vascular cell adhesion molecule 1 (VCAM-1) and fibronectin on endothelial cells and extracellular matrix supports the trafficking toward the BMM[17,18].While CML LSCs have normal expression patterns of VLA-4 and VLA-5,their impaired function demonstrates that these cells are not entirely contingent on β1-integrins for the homing[19].Simultaneously,it has been reported that E and L-selectin and related ligands such as CD44 seem to be closely involved in the bone marrow lodgment of CML LSCs and are considered as the compensatory mechanisms as opposed to normal stem cells[20].Meanwhile,imatinib,which is in first-line therapy for CML,increases another adhesion molecule Ncadherin in CML LSCs.Enhancement of N-cadherin promotes attachment to mesenchymal stromal cells and leads to N-cadherin-β catenin interaction[21].Also,secretion of exogenous WNT by mesenchymal stromal cells activates WNT-β catenin pathway in CML LSCs[21].WNT-β catenin is the leading signaling cascade in selfrenewal and maintenance of normal HSCs and also CML LSCs,and it is important in leukemogenesis and drug resistance[22,23].Although TKIs may attenuate the constative activity of this cascade by targeting P210BCRABL1and destabilize β catenin[24],activationviathe BMM may negate this inhibitory effect.
Apart from direct contacts of CML LSCs with the BMM,secretion of some soluble factors prepares a proper context for the growth of CML LSCs and confers a number of disadvantages for the growth of the normal compartment.It has been reported that enhanced secretion of some chemokines and cytokines,such as macrophage inflammatory protein 1 alpha (MIP-1α) ,MIP-1β,interleukin- 1 alpha (IL-1α),IL-1β,and tumor necrosis factor alpha (TNFα) in the CML BMM,selectively impedes growth of normal HSCs and supports the growth of CML LSCs[15].Furthermore,secretion of IL-10,transforming growth factor beta (TGF-β),and IL-4 by the BMM or by CML LSCs in an autocrine manner downregulates expression of major histocompatibility complex-II (MHC-II) and helps CML LSCs to evade from the immune system and subsequent eradication[25].
A study reported that the higher expression of bone morphogenetic protein receptor type 1b in TKI resistant CML LSCs is activated by bone morphogenetic protein 4viaparacrine and autocrine loops and triggers upregulation of twist family BHLH transcription factor 1,which promotes TKI resistance[26,27].Moreover,paracrine secretion of fibroblast growth factor 2 (FGF2) by mesenchymal stromal cells can provoke imatinib-resistance in CML patients[28].Direct contact of CML cells with mesenchymal stromal cells stimulates secretion of placental growth factor,which in turn increases proliferation and metabolism of leukemic cells and promotes angiogenesis within the BMM[29].
Another secretory factor that reinforces quiescence and resistance of CML LSCs is germane to miR-126.miR-126 is considered to be the regulator of dormancy of CML LSCs as well as of normal HSCs[30].P210BCRABL1kinase activity induces phosphorylation of Sprouty-related,EVH1 domain-containing protein 1,which causes reduction of mature miR-126 in CML LSCs.This depletion should be compensated by an external resource to keep up stemness features[30].In the BMM,endosteal Sca-1+endothelial cells are the credible alternative by providing a high amount of miR-126 possibly through extracellular vesicles[30].Considering this,constraining the activity of miR-126 sensitizes LSCs to TKI and may expedite their removal[30].
Another experiment highlighted the role of the hypoxic BMM in favor of p210BCRABL1independent mechanisms in the survival of CML LSCs.In this milieu,a specific selection of LSC population occurs following the suppression of mature cells and stimulates TKI resistance.Sensitivity of leukemic cells to TKI is rescued by enhanced protein levels of BCRABL1 when LSCs migrate to normoxic condition[31,32].As HSCs reside in the hypoxic endosteal niche,enhancement of low oxygen area in the bone marrow of leukemia patients coincides with resistance and presence of minimal residual disease[33,34].Furthermore,it was demonstrated that hypoxia stabilizes hypoxia-inducible factor1 (HIF1),a transcription factor with a vital role in regulating proliferation,maintenance,and survival of CML LSCs[15].Our knowledge about the interactions of CML LSC with the putative BMM is limited and much remains to be elucidated.Interaction of CML LSCs with their environment through different molecules is described in Table 1 and Figure 2.
AML LSCs AND BONE MARROW MICROENVIRONMENT
Recent studies reported that AML LSCs are highly dependent to the leukemic BMM.In vivocell tracking has specified the anatomical adjacent of these cells to the trabecular osteoblastsviacell adhesion molecules[35].Upregulation of VLA-4 in AML LSCs and its interaction with fibronectin that is distributed abundantly in endosteum facilitates AML LSCs homing to the niche.VLA-4 also has an integrity to VCAM-1 that is expressed by most of the niche cells,particularly endothelial cells[36].These interactions promote drug resistance in LSCs,so that the combination of cytarabine with the antibody against VLA-4 in non-obese diabetic/severe combined immunodeficiency mice prevents AML LSC lodgment to the niche and makes them an easy target[37].Meanwhile,similar to CML LSCs,elevated expression of CD44 on AML LSCs and high hyaluronic acid as its ligand on endosteal niche shift LSCs toward the BMM and chemoresistance state.Furthermore,this interaction promotesactivation of tyrosine kinases and proto-oncogenic signals in leukemic cells including human epidermal growth factor receptor 2,non-receptor kinase Src,Rho-associated protein kinase,and Rac family small GTPase 1[38].While several adhesion molecules and stromal factors are involved in leukemic cell protection in the BMM,the principal mediator is related to the CXCL12-CXCR4 axis[39].Elevated CXCR4 level in AML cells is concomitant with a poor prognosis and causes strong adhesion of AML LSCs to the BMM[40,41].These cells play a bidirectional role by remarked Jagged1 expression that commences Notch1 pathway in neighbor leukemic cells and promotes autocrine signals in Jagged1 expressed stromal cells within the niche.Activation of Notch1 pathway accelerates self-renewal capacity of LSCs[42,43].
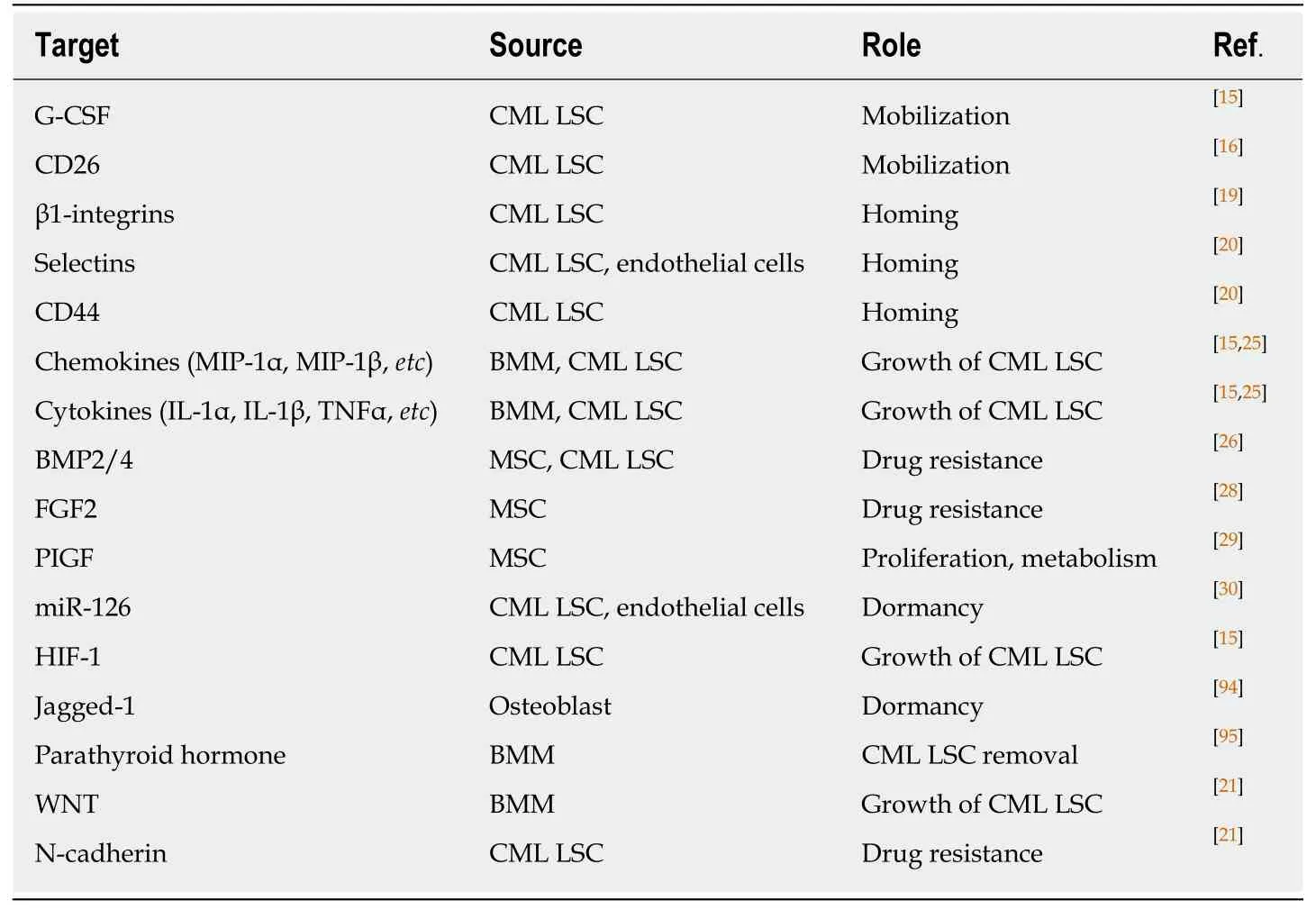
Table1 Possible molecules and their role in chronic myeloid leukemia stem cells-bone marrow microenvironment interaction
When AML LSCs reside in this supportive milieu,secretion of some growth factors,cytokines,and chemokines is considerably important to keep leukemogenesis up in the BMM.Secretion of IL-8 in an autocrine mechanism and its receptor CXCR2 by AML LSCs supports IL8-CXCR2 interaction and triggers activation of multiple pathways,including PI3K/AKT,phospholipase C/protein kinase C,mitogenactivated protein kinase,β catenin,HIF-1,and nuclear factor kappa-light-chainenhancer of activated B cells (NF-kB) in AML LSCs that brings about tumor progression and survival[44].Moreover,CXCR2 inhibition reverses the growth of AML LSCs and enhances their removal[44].
Another study reported that elevated parathyroid hormone signaling in osteoblastic cells controls HSC pool.While parathyroid administration increases the number of AML LSCs,it decreases the number of CML LSCs and reflects the distinct role of the BMM components in different hematologic malignancies[45].Activation of angiopoietin-Tie2 signaling in the osteoblastic niche preserves AML LSCs in dormancy and stimulates drug resistance[46].Meanwhile,release of pro-angiogenesis factors,such as vascular endothelial growth factor,hepatocyte growth factor,basic fibroblast growth factor,TNFα,and vascular endothelial growth factor receptor by LSCs increases neoangiogenesis.On the other hand,secretion of inflammatory and proliferative cytokines like TNFα,IL-6,IL-1β,and G-CSF by leukemic cells and granulocyte-monocyte CSF by endothelial cells shares in niche neo vasculature that is considered as the major foundation of leukemia progression by providing metabolites and oxygen for AML LSC[47-51].In some conditions,human AML LSCs increase vascular permeability to reduce nitric oxide levels produced during the anaerobic glycolytic pathway[52].In a close relationship,endothelial cells also mediate proliferation and survival of LSCs by elevating the expression of CXCR4[53].

Figure2 CML LSCs and their interaction with the bone marrow microenvironment.
AML LSCs are capable of maintaining long term reconstitution in the hypoxia environment and modulate the differentiation process[54].This finding is in agreement with low metabolism and energy status of AML LSCs in the BMM.However,during stresses and apoptosis,high expression of CD36,a fatty acid transporter,and enhanced lipolysis by leukemic stem cells provide a compensatory source of energy that underlies their persistence[55,56].More investigations in LSCs and BMM crosstalk are needed to provide new insights to leukemogenesis biology and effective strategies for leukemia treatment.Interaction of AML LSCs with their environment through different molecules is summarized in Table 2 and Figure 3.
SPECIFIC MARKERS OF CML AND AML STEM CELLS
As CML LSCs reside in the CD34+/CD38- cell fraction,finding specific markers is one step ahead for recognizing and selectively targeting these cells and to discriminate from normal HSCs.A useful CD marker should first distinguish between normal and leukemic stem cells,and,second,show lack or limited expression on the more mature population.
Many markers,such as CD44 and CD117[57,58],have been recommended for detection of CML LSC,but shared expression with normal HSC has limited their application.On the other hand,surface markers such as CD25,IL-1 receptor accessory protein (IL-1RAP),and CD26 may offer a viable alternative in segregating CML LSCs[16,59,60].CD25 (IL2Rα),which is expressed by CML LSCs,is regulated by signal transducer and activator of transcription 5 activity and serves as the suppressor of cell growth in CML LSCs.However,expression on the surface of progenitor cells might also be detectable[59].IL-1RAP as a co-receptor of IL-1 receptor participates in activation of NF-kB and AKT signaling pathways that promote the growth of CML LSCs.As IL-1RAP expression increases with the disease progression,it seems that it may be a diagnostic marker for the advanced phase of the CML[60].CD26,with a chemokine cleavage activity,has a role in the mobilization of the CML LSCs into the blood by cleaving CXCL12[16,61].Expression of this marker in CML is just limited to CML LSCs in the chronic phase and is not expressed by normal HSCs,more mature population,and acute phase of the disease.So,CD26 may be regarded as a targetmarker for detection of CML LSCs in newly diagnosed patients[16].While acute lymphoblastic leukemia LSCs with P210BCRABL1also express CD26[62],whether its expression in acute lymphoblastic leukemia and CML LSC is P210BCRABL1dependent or independent remains to be discovered.
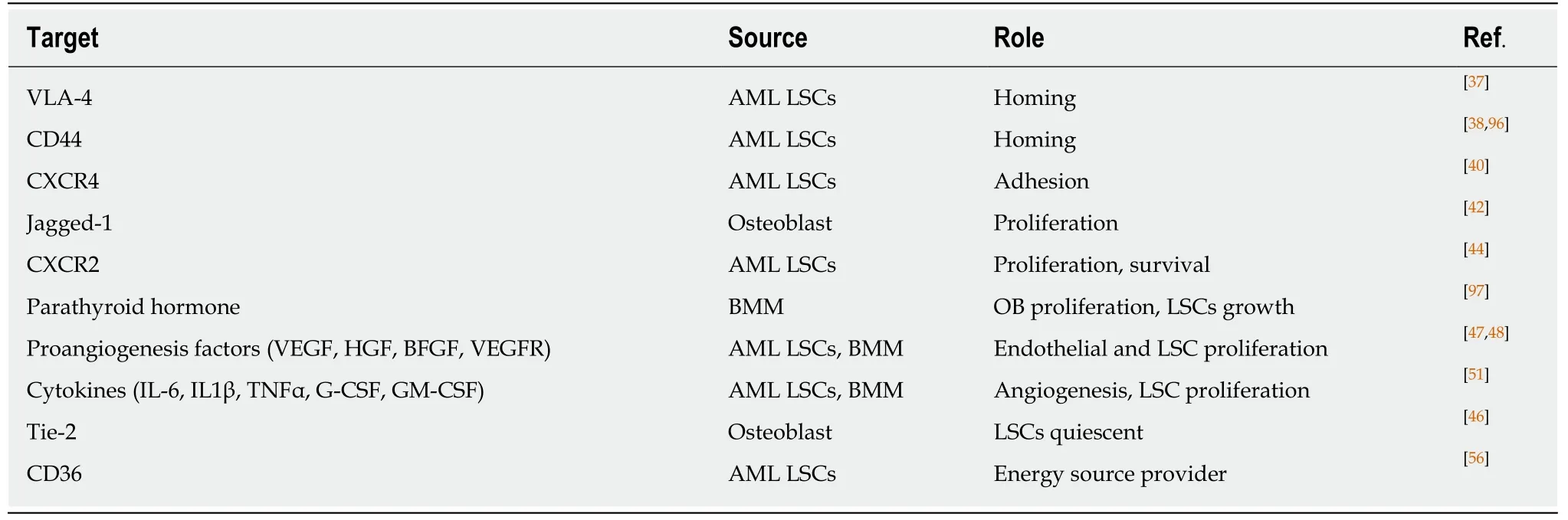
Table2 Possible molecules and their role in acute myeloid leukemia stem cells-bone marrow microenvironment interaction
In contrast to the chronic phase of CML in which CML LSCs are defined in the CD34+/CD38- fraction,AML LSCs are composed of heterogenous populations and except the CD34+/CD38- fraction,they also reside in CD34+/CD38+ and CD34-fractions[63,64].While the preleukemic state in AML initiates in HSC,they are considered non-leukemic,and progenitors are responsible for leukemia development.It has been reported that lymphoid primed multipotent progenitor cells in CD34+/CD38- fraction and granulocyte-macrophage progenitors in CD34+/CD38+fraction are major AML LSC populations and that lymphoid primed multipotent progenitor cell like cells give rise to granulocyte-macrophage progenitor like cells (not vice versa) and show a higher self-renewal capability[63].However,based on the engraftment potential and transcriptomic analysis,CD34 is not a determinant marker of AML LSCs,and other markers are needed for the identification of these cells.Meanwhile,CML acute phase mimics the same pattern as acute leukemia,and LSC populations in acute phase of CML are extended to different types of progenitor cells that reflect LSCs heterogeneity[65].So,considering these,finding a proper marker to differentiate normal and leukemic stem cells in AML seems rather difficult and applying different markers is indispensable.For instance,some markers,such as CD96[66],C-type lectin-like molecule-1[67],CD123[68],CD25[69],CD47[70],T-cell immunoglobulin and mucin domain-3[71],etc,have been proposed for AML LSCs and are variably expressed by AML patients.In this case,a panel of markers might be helpful in dealing with AML LSCs.Apart from diagnosis,targeting of CML and AML LSCs based on these markers is already well underway,which may open up an opportunity to eliminate selectively LSCs and spare normal stem/progenitor cells.Different markers proposed for CML and AML LSCs are summarized in Table 3 and Figure 4.
TARGETING LEUKEMIC STEM CELLS AND THEIR ENVIRONMENT
Clinical trials have been reported that about 40%-60% of CML patients are eligible for treatment discontinuation[72,73].While losing MR3in CML patients is considered the sign of TFR failure,almost all of them achieve major molecular response and deeper molecular responses after resuming the treatment[74,75].Identification of the minimal residual disease is dependent on the application of quantitative real-time polymerase chain reaction,and subsequently it has been confirmed that CML LSCs are present from diagnosis,during the treatment and also in patients who are in TFR.These cells may be considered BCRABL1 negative due to undetectable transcript level of BCRABL1 in CML LSCs[76].Furthermore,an inverse correlation between the number of residual CD26+ CML LSCs and the probability of remaining in TFR has been reported[76].Whereas CML LSCs are insensitive to common TKI therapy,targeting the BMM and breaking the close intimacy between CML LSCs and the BMM may help more patients achieve TFR and sustain it for a longer period.
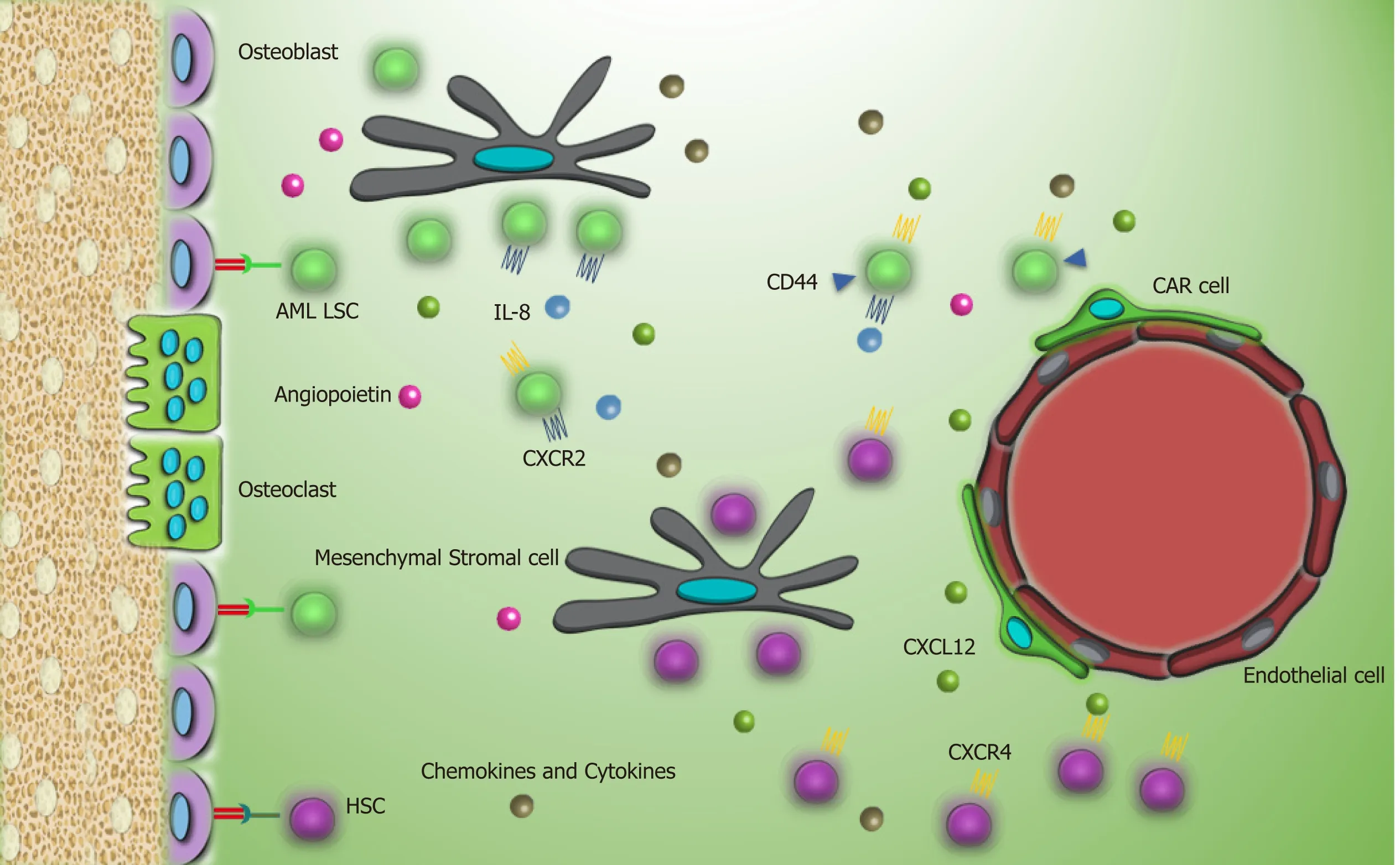
Figure3 AML LSCs and their interaction with the bone marrow microenvironment in contrast to chronic myeloid leukemia stem cells,AML LSCs have high expression of CXCR4 that help them to reside in the bone marrow microenvironment.
A promising option in targeting CML LSCs is to disrupt the connection between these cells and the BMM,making them more sensitized to conventional therapy.Since the presence of CXCR4-CXCL12 axis enhances proliferation and survival of CML cells by upregulation of different signaling pathways,such as extracellular signalregulated protein kinases 1 and 2,AKT,and Janus kinase (JAK)/STAT,interrupting this axis may dwindle the protective role of the BMM[77,78].It has been reported that plerixafor (AMD31000),a CXCR4 antagonist,in combination with different generations of TKIs failed to reduce residual disease burden[79].However,another experiment proved the potent role of BKT140,another antagonist of CXCR4,in declining the growth of leukemic cells bothin vitroandin vivo[77].
IL-1RAP is a good marker for targeting CML LSCs in a selective manner due to its specific expression on CML LSCs.It was reported using an antibody against IL-1RAP that IL-1RAP potentially targets CML LSCs while normal stem cells remain untouched[80].This killing effect was increased when TKIs were used in combination[80].However,the limitation of therapeutic antibodies led to the introduction of IL1RAP CAR T cell,which is a prominent approach in dealing with resistant CML LSCs[81].
As mentioned above,secretion of some cytokinesviaautocrine or paracrine mechanisms helps CML LSC to escape from the immune system.These cytokines proceed through activation of JAK,which may activate in a P210BCRABL1independent fashion.So,applying ruxolitinib,a JAK inhibitor,might help upregulate MHC-II expression in CML LSCs and increase their immunogenicity for the detection and targeting by the immune system[25,82].
While targeting AML LSCs as leukemia-initiating cells may guarantee duration of the remission,eradication of these cells seems difficult because of their heterogeneity.In targeting AML LSCs,we have a vast variety of options considering cell cycle,surface markers that are useful for the segregation from normal HSCs,oncoproteins,and epigenetic participants[83].However,the supportive role of the BMM is an undeniable fact and affects all pathways related to cell protection.Therefore,combination therapy with specific targets in the BMM is a promising approach toovercome resistance and to eradicate LSCs more effectively[10,84].

Table3 Chronic myeloid leukemia and acute myeloid leukemia stem cell markers for detection and selective targeting
It was reported that blocking CXCR4 by plerixafor suppresses CXCL12-CXCR4 axis and increases the release of AML LSCs from the bone marrow to the blood[85].AMD3465,another CXCR4 antagonist,in combination with G-CSF and bortezomib,a proteasome inhibitor,prevents AML LSC migration toward the BMM and consequently makes them more accessible to chemotherapy agents[86,87].Meanwhile,in the leukemic BMM,HIF1-α and vascular endothelial growth factor modulate expression of CXCR4 and CXCL12,and targeting of these two in combination with CXCR4 antagonists significantly reduces homing of myeloid leukemia cells and reflects inducing mobilization of these cells to the blood might suppress leukemia development[88].
On the other hand,upregulation of CD44,VLA-4,and Tie2 on AML LSCs is considered a putative target.Anti-CD44 therapy in AMLs prevents LSCs homing.Also,neutralizing VLA-4 antibody together with cytarabine treatment hampers AML development in a patient-derived xenograft mouse model[38,89].Adhesion of LSCs to mesenchymal stromal cellsviaVLA-4/VCAM-1 axis triggers NF-kB activation as an anti-apoptotic factor in AML LSCs and stromal cells.AS101,a VLA-4 inhibitor that is in Phase II of a clinical trial,prevents NF-kB activation and renders LSCs to chemotherapy[90].Whilst interaction of Tie2 with Ang-1 concludes LSCs quiescent,disruption of Ang-1/Tie2 interaction makes cells to cycle and recover LSCs sensitivity to cell cycle targeting agents.Ang1/2 neutralizing peptibody Trebananib (AMG 386),a combination of a peptide with an antibody,demonstrated promising results in a monotherapy program in a clinical trial[91].Another putative marker in AML LSCs is CD47 (SIRPα ligand),which is highly expressed by these cells.Interaction of CD47 with its ligand blocks phagocytosis,while blockade of this molecule leads to tumor cell phagocytosis and AML LSCs elimination in an efficient manner[70].Direct contact of AML LSCs with the BMMviaNotch1-Jagged interaction initiates Notch signaling by intracellular domain cleavage of Notch1 following -secretase activation.Application of -secretase inhibitors like dibenzazepine in order to inhibit Notch signaling culminates in the suppression of LSC cell growth[83].However,in Kannanet al[92],a pan-Notch inhibitor could not affect LSC proliferation,which confirms further study is needed to consider Notch signaling for targeting AML LSCs.
Inducing apoptosis also is a common approach in AML targeted therapy.O' Reillyet al[93]reported that microenvironment mediated drug resistance in AML might occur following overexpression of myeloid cell leukemia 1,a BCL-2 family protein,in mesenchymal stromal cells.They confirmed that inhibition of myeloid cell leukemia 1 reverts the BMM mediated resistance against cytarabine and daunorubicin,prevents disease relapse,and ultimately improves patient survival.The proposed compounds under clinical trials related to targeting CML and AML LSCs interaction with BMM are summarized in Table 4.Other studies reported another possible target for elimination of AML LSCs by inhibiting the IL8-CXCR2 axis.This approach selectively eliminates AML LSCs while sparing normal HSCs[44].
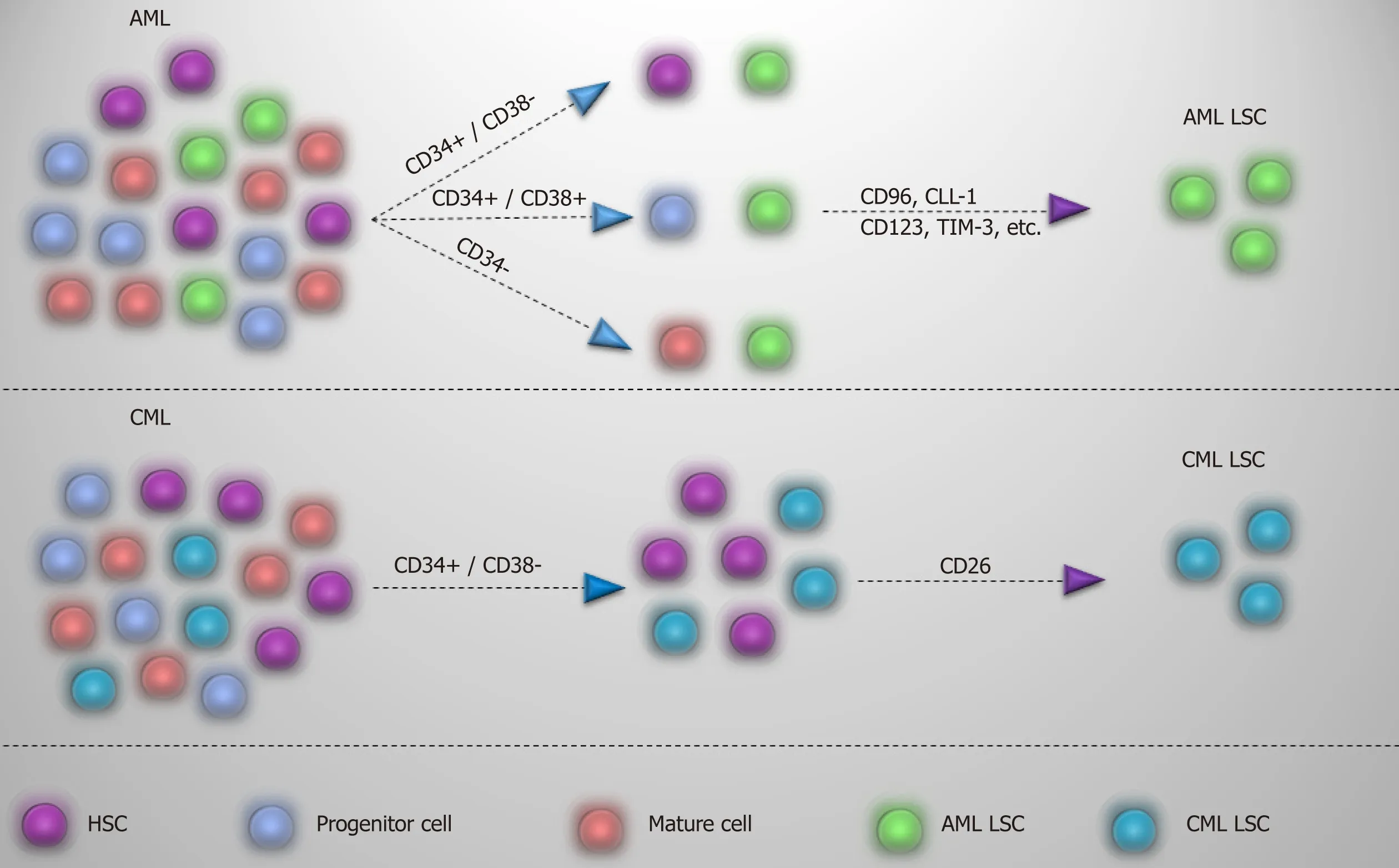
Figure4 Detection of AML and CML LSCs.
CONCLUSION
The therapeutic approaches that we listed above are in most cases already the object of investigational clinical trials.Many others will certainly follow,and,as far as our knowledge about the biology,the phenotypical appearance and the biochemical pathways typical of the leukemic stem cells will be better understood.It is unlikely that a single agent will be able to eliminate the leukemic stem cells.Targeted therapy will most likely be a combination of new drugs and more conventional therapeutic agents,ranging from traditional chemotherapy to new molecularly targeted agents or immune modulating agents.The final goal that we hope to achieve is to cure the vast majority of our patients and to improve their quality of their life.
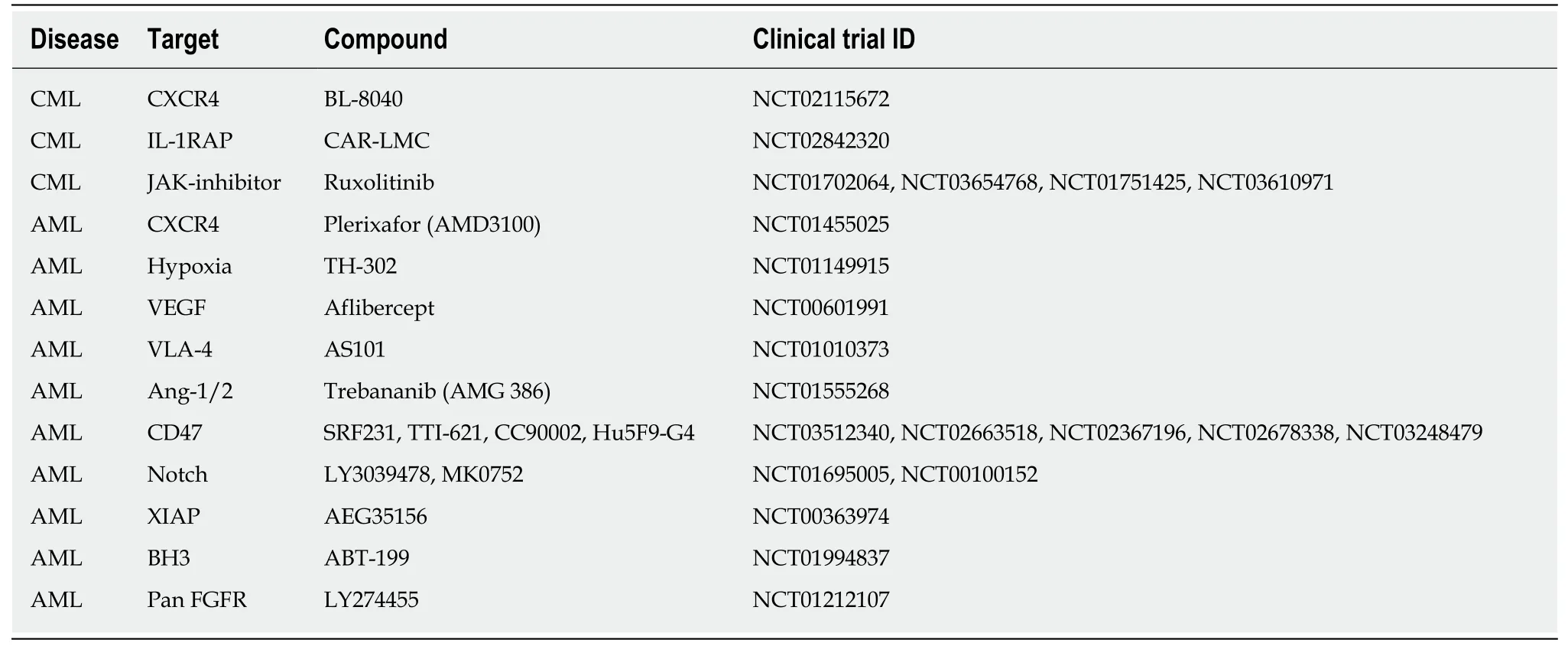
Table4 A draft of compounds under clinical trial in leukemic stem cell bone marrow microenvironment target therapy
 World Journal of Stem Cells2019年8期
World Journal of Stem Cells2019年8期
- World Journal of Stem Cells的其它文章
- Moving forward on the pathway of cell-based therapies in ischemic heart disease and heart failure - time for new recommendations?
- Neural regeneration by regionally induced stem cells within poststroke brains:Novel therapy perspectives for stroke patients
- Orchestrating stem cell fate:Novel tools for regenerative medicine
- Tonsil-derived stem cells as a new source of adult stem cells
- Linking stemness with colorectal cancer initiation,progression,and therapy
- Derivation and applications of human hepatocyte-like cells