
高效液相色谱-串联质谱法测定奶粉中氯酸盐和高氯酸盐
2019-09-16 08:22周晓晴吕小丽万建春席慧婷
色谱 2019年10期
周晓晴, 吕小丽, 万建春, 郭 平*, 郭 丹, 席慧婷
(1. 江西省食品检验检测研究院, 江西 南昌 330001;2. 南昌大学食品科学与技术国家重点实验室, 江西 南昌 330047)
氯酸盐和高氯酸盐是一类生活中普遍存在的有害污染物。氯酸盐是二氧化氯消毒产生的无机副产物,也可由自然界中含氯化合物分解产生。氯酸盐具有强氧化性,会影响人体的血液系统,引起高铁性血红蛋白血症[1]和贫血症,也可能导致神经和呼吸道中毒,降低精子活力和数量[2-4]。高氯酸盐常用作化肥原料,大气中也能够产生高氯酸根[5]。人工合成的高氯酸盐作为氧化剂被广泛用于烟花生产、橡胶制造、皮革加工、火箭固体推进剂等化工领域[6],生产中不合理的处理易导致其迁移至地表水中,污染土壤、饮用水和食品。毒理学研究[7,8]表明,高氯酸盐对人体健康的影响主要集中在甲状腺功能方面,它可抑制碘的吸收,引起甲状腺功能失调,影响人体正常生长发育。考虑健康风险,欧洲食品安全局(EFSA)设定氯酸盐和高氯酸盐的每日可耐受摄入量分别为3和0.3 μg/kg BW/day[9]。在奶粉生产过程中,氯酸盐和高氯酸盐可能作为中间生产的污染物,残留在奶粉中,而我国目前暂未制定氯酸盐和高氯酸盐的限量标准,因此对奶粉中的残留量进行监控十分必要。
近年来,应用于检测氯酸盐和高氯酸盐的方法有分光光度法[10]、离子色谱法[11,12]、离子色谱-质谱联用技术[13-15]、高效液相色谱-质谱/质谱法等[16-18]。分光光度法的检测设备条件要求不高,早期用于高氯酸盐含量的粗筛,由于设备灵敏度的问题,只适用于较高含量的测定。离子色谱虽能对饮用水、水果和蔬菜等各种食品进行较为准确地定量,但电导检测器缺乏选择性,离子色谱法存在干扰严重、检测易假阳性和不能进行痕量定量的缺点。离子色谱-质谱虽较离子色谱有很大优势,但IC-MS/MS在实际应用中存在普及率低、离子色谱柱不耐受有机溶剂、大体积进样易造成柱子过载等问题。随着液相色谱-质谱联用技术的发展,这种技术灵敏度高、定性准确,正越来越广泛地应用在了高氯酸盐的检测分析中。但应用液相色谱-质谱联用技术同时测定奶粉中氯酸盐和高氯酸盐的文献报道相对较少。
奶粉基质复杂,为提高氯酸盐和高氯酸盐检测的准确性,本实验在已有研究基础上,利用LC-MS/MS结合同位素内标法定量检测手段,建立了奶粉中氯酸盐和高氯酸盐的测定方法。与现有的高效液相色谱-串联质谱法[16-18]相比,通过优化样品提取剂和固相萃取净化过程,选择0.1%(v/v)甲酸-乙腈对样品进行提取,提取液经PRiME HLB固相萃取柱净化后直接上机分析,无需浓缩,操作简单,准确度高,改善了谱峰宽的不足。本方法可适用于奶粉等乳制品中氯酸盐和高氯酸盐的快速检测和监控。
1 实验部分
1.1 仪器、试剂与材料
ACQUITY UPLC TQS高效液相色谱-质谱仪(配备电喷雾离子(ESI)源)(美国Waters公司); G-16离心机(德国Sartorius公司); Vortex Wizard涡旋仪(意大利VELP公司); VAC ELUT-20固相萃取装置(美国Agilent公司); Synergy纯水系统(德国Merck Millipore公司)。Thermo Scientific Acclaim TRINITY P1复合离子交换柱(50 mm×2.1 mm, 3 μm)(美国赛默飞公司)。MCE混合纤维素酯膜(0.22 μm,岛津技迩(上海)商贸有限公司); PES聚醚砜膜(0.22 μm,津腾);尼龙膜(0.22 μm,岛津技迩(上海)商贸有限公司)。PRiME HLB、C18和PSA(N-丙基乙二胺)固相萃取柱(美国Waters公司);石墨化炭黑Carb和OnGuard Ⅱ RP固相萃取柱(博纳艾杰尔科技有限公司)。氯酸盐标准品溶液(1 000 mg/L,农业部环境保护科研监测所),18O3氯酸盐同位素内标(200 mg/L,美国EURL-SRM公司),高氯酸钠标准品溶液、18O4高氯酸盐同位素内标(均为100 mg/L,美国CIL公司);甲醇、乙腈均为色谱纯(西班牙Scharlau公司),甲酸、乙酸铵为色谱纯(上海阿拉丁生化科技股份有限公司);实验用奶粉样品均购自超市。
1.2 标准溶液的配制
氯酸盐标准中间溶液:准确吸取0.1 mL氯酸盐标准品溶液(1 000 mg/L)于10 mL容量瓶中,用超纯水稀释至刻度,摇匀,制成质量浓度为10 mg/L的氯酸盐标准中间溶液,置于4 ℃冰箱保存。
混合标准中间液:分别准确吸取2 mL氯酸盐标准中间溶液(10 mg/L)、0.1 mL高氯酸钠标准品溶液(100 mg/L),置于同一10 mL容量瓶中,用超纯水稀释至刻度,摇匀,制成氯酸盐、高氯酸盐质量浓度分别为2.0、1.0 mg/L的混合标准中间液,置于4 ℃冰箱保存。
混合同位素内标液:分别准确吸取0.05 mL氯酸盐同位素内标(200 mg/L)、0.1 mL高氯酸盐同位素内标(100 mg/L),置于同一10.0 mL容量瓶中,用超纯水稀释至刻度,摇匀,制成氯酸盐-18O3、高氯酸盐-18O4质量浓度分别为4.0、1.0 mg/L的混合同位素内标液,置于4 ℃冰箱保存。
混合标准曲线的配制:分别准确吸取混合标准中间液及混合同位素内标液适量,用20 mmol/L乙酸铵-甲醇溶液(1∶2, v/v)稀释,制成系列质量浓度的氯酸盐溶液:2.0、4.0、10.0、20.0、40.0 μg/L,和高氯酸盐溶液:1.0、2.0、5.0、10.0、20.0 μg/L。混合标准工作液中,氯酸盐-18O3、高氯酸盐-18O4的质量浓度分别为20.0、5.0 μg/L。
1.3 样品前处理
1.3.1样品提取
准确称取试样2 g(精确至0.001 g),置于50 mL具塞离心管中,加入75 μL混合同位素内标液,再加入5 mL 0.1%(v/v)甲酸水溶液,迅速混匀,置于45 ℃水浴超声20 min,涡旋振荡5 min,再准确加入10.0 mL乙腈,混匀,于10 000 r/min下常温离心10 min,取上清液待净化。
1.3.2样品净化
取3 mL上清液过PRiME HLB固相萃取柱,并接上0.22 μm MCE混合纤维素酯膜,收集续滤液,供液相色谱-串联质谱仪测定。
1.4 分析条件
1.4.1色谱条件
色谱柱:Thermo Scientific Acclaim TRINITY P1复合离子交换柱(50 mm×2.1 mm, 3 μm)(美国赛默飞公司);柱温:35 ℃;流动相:A为乙腈,B为20 mmol/L乙酸铵溶液;流速:0.5 mL/min。梯度洗脱程序:0~0.5 min, 35%A; 0.5~4.0 min, 35%A~65%A; 4.0~5.0 min, 65%A~90%A; 5.0~7.0 min, 90%A; 7.0~8.0 min, 90%A~35%A; 8~10 min, 35%A。进样量:3 μL。
1.4.2质谱条件
离子源:ESI源,负离子模式;多反应监测(MRM)模式;毛细管电压:3 kV;锥孔电压:60 V;脱溶剂温度:600 ℃;脱溶剂气流量:1 000 L/h;锥孔气流量:150 L/h。4种化合物的其他质谱参数见表1。
2 结果与讨论
2.1 质谱条件的选择
以氯酸盐标准溶液为对象,在负离子模式下,用全扫方式进行扫描,确定目标物的分子离子,考虑子离子丰度很弱,为满足低分辨质谱四分法原则,选取两对母离子m/z82.9和m/z84.9,在一定碰撞气和碰撞能条件下通过二级质谱碎裂,分别获取了子离子m/z66.9和m/z68.9,选取m/z66.9为定量离子。按同样的方法得到高氯酸盐的母离子为m/z98.9和m/z100.9,子离子m/z83.0和m/z85.0,选取m/z83.0为定量离子。根据氯酸盐和高氯酸盐的响应手动调节确定锥孔电压为60 V,优化后的碰撞能量参数见表1。

表 1 氯酸盐和高氯酸盐的母离子、子离子、碰撞能量及内标物
* Quantitative ion.
2.2 色谱柱的选择
氯酸盐和高氯酸盐均为具有极性的离子化合物,在反相色谱柱C18柱上难以保留,易出现峰形宽、拖尾或分叉的现象。SN/T 4089-2015[19]中使用IC-PakTMAnion HR的一款阴离子交换型色谱柱,高氯酸盐峰形宽,时间跨度为1 min。Thermo Scientific Acclaim TRINITY P1柱属于离子交换型色谱柱,其填料的键合相可提供阴阳离子交换和反相的功能。本方法选取Thermo Scientific Acclaim TRINITY P1柱进行测定。考虑色谱柱洗脱需用盐类,故选择液相色谱-串联质谱法中较常用且较易挥发的乙酸铵。Thermo Scientific Acclaim TRINITY P1柱兼具反相功能,洗脱需用有机溶剂,乙腈洗脱能力大于甲醇,综合考虑选择20 mmol/L乙酸铵溶液-乙腈为流动相,流速0.5 mL/min。在此条件下,氯酸盐出峰时间为1.58min,高氯酸盐出峰时间为3.84min。氯酸盐和高氯酸盐标准品的MRM色谱图见图1。
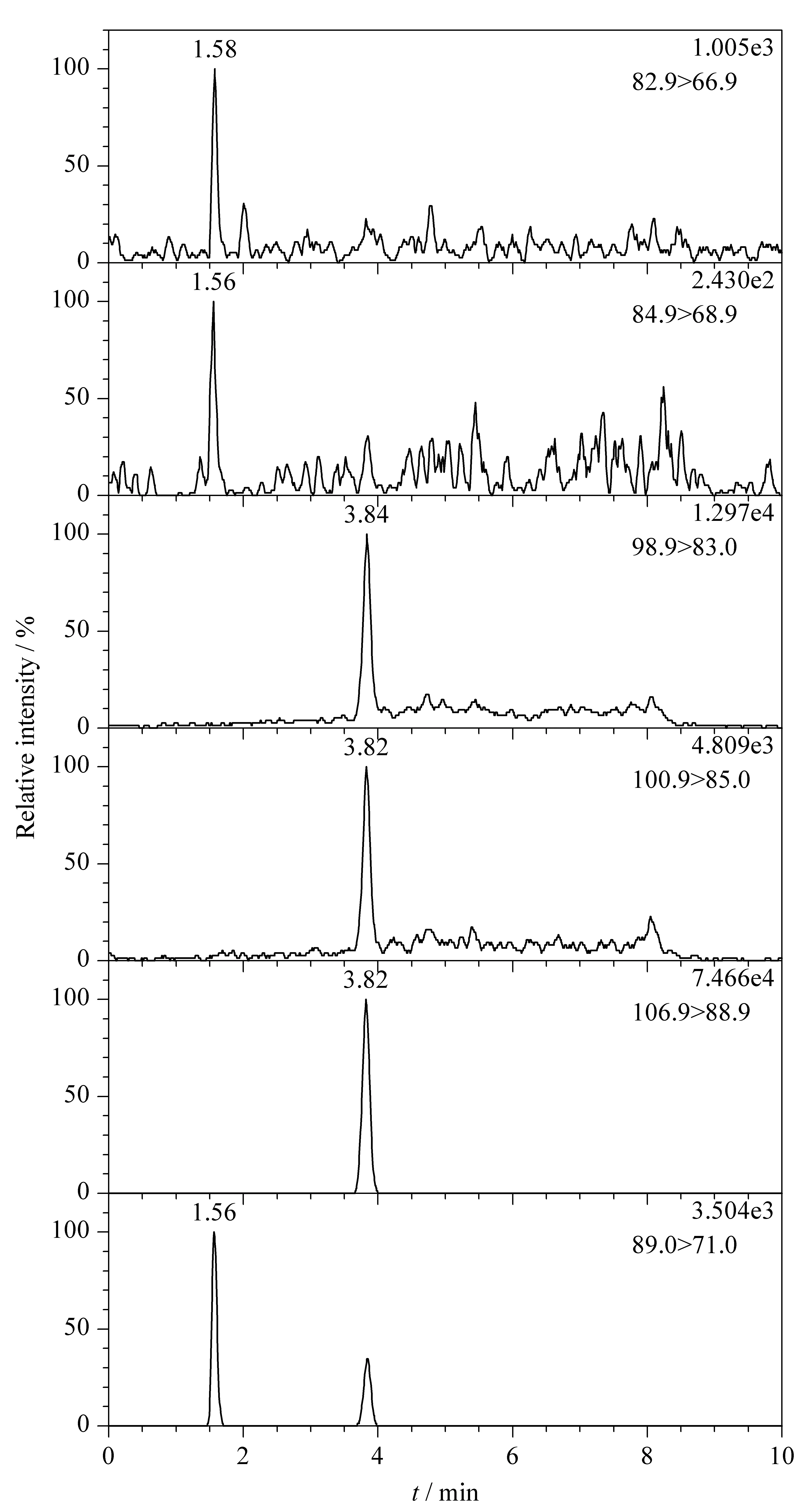
图 1 氯酸盐、高氯酸盐及其内标的MRM色谱图Fig. 1 Multiple reaction monitoring (MRM) chromatograms of chlorate, perchlorate and their internal standards
2.3 前处理方法的优化
2.3.1滤膜的选择
在色谱分析中,为避免堵塞管路,通常选用滤膜对待测样液进行过滤。本文比较了MCE混合纤维素酯膜、PES聚醚砜膜和尼龙膜对氯酸盐和高氯酸盐测定的影响。用0.1%(v/v)甲酸水-乙腈(1∶2, v/v)配制10 μg/L的混合标准溶液,分别过MCE混合纤维素酯膜、PES聚醚砜膜和尼龙膜后,与未过膜的10 μg/L混合标准溶液一同上机检测,同时做滤膜空白试验。
3种滤膜的不加标准溶液的空白试验均未检出目标组分。从表2实验结果可以看出,过膜对4种化合物的回收率有影响。其中尼龙膜的影响最大,使高氯酸盐和高氯酸盐内标的回收率下降近约90%,说明滤膜会对目标物有吸附,所以导致回收率下降。结果表明,尼龙膜的吸附作用最强,PES聚醚砜膜次之,尼龙膜和PES聚醚砜膜的吸附情况与吴映璇等[20]的研究结果一致。因此最终确定采用MCE混合纤维素酯膜,然后上机检测。

表 2 不同滤膜对氯酸盐和高氯酸盐的吸附情况
2.3.2净化柱的选择
固相萃取柱是净化的主要方式,本文比较了PRiME HLB固相萃取柱、C18固相萃取柱、石墨化炭黑Carb固相萃取柱、PSA固相萃取柱和OnGuard Ⅱ RP固相萃取柱对氯酸盐和高氯酸盐及其内标的净化效果。
使用前,C18固相萃取柱分别用5 mL色谱纯甲醇和5 mL超纯水进行活化,石墨化炭黑Carb固相萃取柱、PSA固相萃取柱和OnGuard Ⅱ RP固相萃取柱分别用5 mL色谱纯甲醇进行活化。分别在准备好的5种固相萃取柱上上样5 mL 10 μg/L的氯酸盐和高氯酸盐混合标准溶液,收集全部流出液与未过柱的10 μg/L混合标准溶液一同上机检测,实验结果如表3所示。
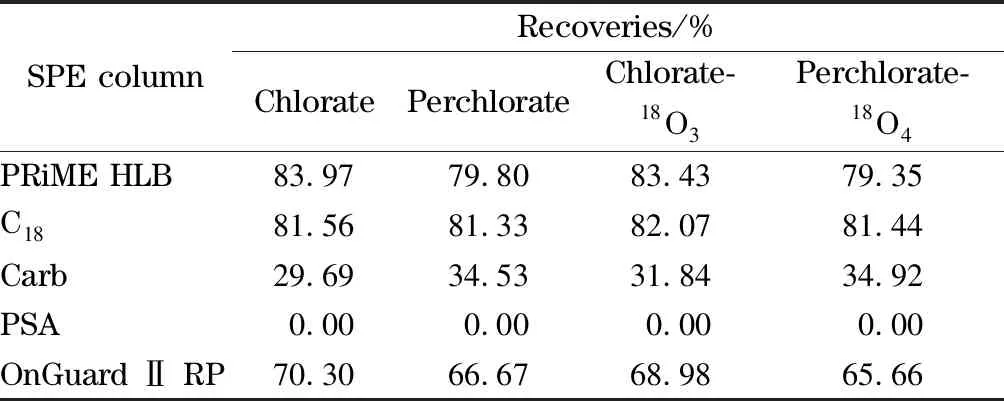
表 3 经5种固相萃取柱净化后4种化合物的回收率
实验结果显示,PRiME HLB固相萃取柱和C18固相萃取柱的回收率相近,优于OnGuard Ⅱ RP固相萃取柱和石墨化炭黑Carb固相萃取柱,PSA固相萃取柱的回收率最差。由于PRiME HLB固相萃取柱不用活化,具有操作时间短、节省试剂的优点,本方法最终选取PRiME HLB固相萃取柱为净化柱。
2.3.3提取溶剂的选择
称取2 g奶粉样品。氯酸盐和高氯酸盐的添加量分别为60和30 μg/kg。分别选用超纯水-甲醇(1∶2, v/v)、超纯水-乙腈(1∶2, v/v)、0.1%甲酸水溶液-甲醇(1∶2, v/v)、0.1%甲酸水溶液-乙腈(1∶2, v/v)、0.05%氨水溶液-甲醇(1∶2, v/v)、0.05%氨水溶液-乙腈(1∶2, v/v)进行提取,采用PRiME HLB固相萃取柱净化,计算氯酸盐和高氯酸盐的回收率(见图2)。实验结果表明,使用0.1%甲酸水溶液-乙腈的提取净化效果最优,对氯酸盐和高氯酸盐的回收率稳定(98.06%~100.69%和103.69%~105.86%)。0.1%甲酸水溶液-甲醇、超纯水-乙腈、0.05%氨水溶液-乙腈、超纯水-甲醇进行提取时,存在一定杂质干扰,提取液会出现混浊现象,0.05%氨水溶液-甲醇的提取液无法过柱净化。
奶粉中含有蛋白质、脂肪、碳水化合物等有机成分。乙腈去除蛋白质的能力优于甲醇。相比其他试剂,乙腈-水体系提取出来的脂肪相对较少,而酸性条件有利于蛋白质的沉淀。因此,本方法选择用5 mL 0.1%甲酸水溶液提取20 min后,加10 mL乙腈测定蛋白质。
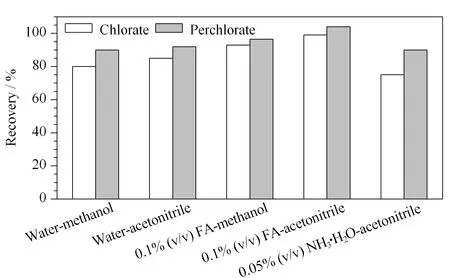
图 2 不同提取溶液对氯酸盐和高氯酸盐回收率的影响Fig. 2 Effect of the different extraction solutions on the recoveries of chlorate and perchlorateFA: formic acid.
2.4 基质效应
采用标准曲线比较法[7],对奶粉前处理方法的基质效应进行评估。标准曲线A:用20 mmol/L乙酸铵-甲醇溶液(1∶2, v/v)稀释,制成含氯酸盐依次为2.0、4.0、10.0、20.0、40.0 μg/L,高氯酸盐依次为1.0、2.0、5.0、10.0、20.0 μg/L的混合标准工作液。标准曲线B:用不含氯酸盐和高氯酸盐的奶粉样品,按照1.3节样品前处理方法进行提取净化,用净化液配制同样质量浓度的系列标准曲线。由基质效应(ME,%)=斜率B曲线/斜率A曲线×100%计算,得到氯酸盐的ME为63%,高氯酸盐的ME为83%。结果显示,奶粉样品的基质效应为基质抑制,因此采用外标法定量存在误差。采用同位素内标法,同位素内标与待测目标物的色谱和质谱行为相近,可以补偿氯酸盐和高氯酸盐因基质效应影响引起的响应变化,以解决因基质效应造成定量不准的问题[3,9,21]。
2.5 方法学验证
2.5.1线性范围和定量限
将不同质量浓度的氯酸盐和高氯酸盐标准溶液按本方法确定的条件进行测定,以氯酸盐和高氯酸盐及其对应同位素内标的峰面积比值为纵坐标,以质量浓度比值为横坐标,绘制标准曲线。结果表明,氯酸盐和高氯酸盐在线性范围内呈良好的线性关系,线性相关系数(r2)均大于0.999(见表4)。以10倍信噪比(S/N)确定氯酸盐的定量限,为15.0 μg/kg,高氯酸盐的定量限为7.5 μg/kg。

表 4 氯酸盐和高氯酸盐的回归方程、相关系数和定量限
ND: not detected.
2.5.2回收率和精密度
选取氯酸盐和高氯酸盐污染水平较低的奶粉样品,参照GB/T 27404-2008[22]在2倍、4倍、8倍方法定量限水平,做加标回收试验和精密度试验。其中氯酸盐的添加水平为30.0、60.0、120.0 μg/kg,高氯酸盐的添加水平为15.0、30.0、60.0 μg/kg,每个水平重复测定6次,测得的回收率和精密度见表5。结果显示,方法的回收率为89.24%~107.85%,相对标准偏差为3.15%~10.42%,符合GB/T 27404-2008标准的要求。实验空白、样品和加标样品的MRM色谱图见图3。
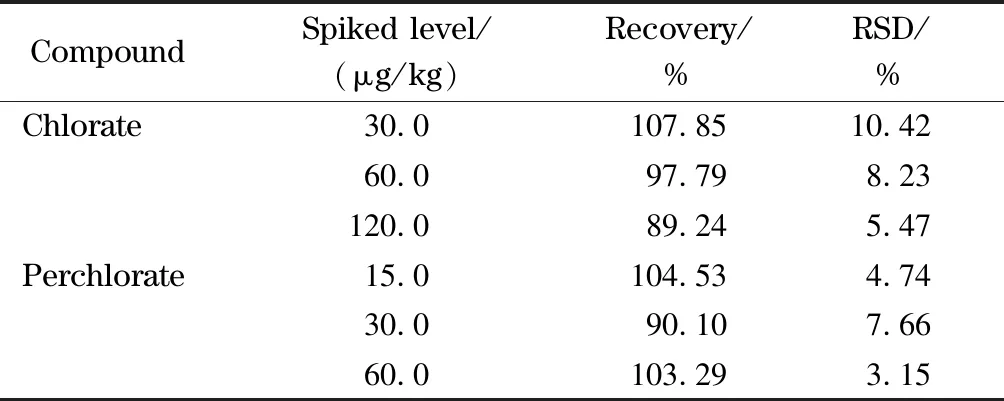
表 5 氯酸盐和高氯酸盐的回收率及精密度(n=6)
2.6 实际样品的测定
用本文建立的方法对实际奶粉样品进行检测。10批奶粉样品的检测结果如表6所示。氯酸盐检出5批,含量范围在19.62~85.25 μg/kg之间。大多数样品中,高氯酸盐均有不同程度的检出,其中6批样品的高氯酸盐含量超出方法定量限7.5 μg/kg,含量范围为10.34~28.79 μg/kg。

图 3 实验空白、样品和加标样品的MRM色谱图Fig. 3 MRM chromatograms of the blank, a sample, and a spiked sampleSpiked levels: chlorate, 60.0 μg/kg; perchlorate, 30.0 μg/kg.

表 6 奶粉样品中氯酸盐和高氯酸盐的测定结果
ND: not detected.
3 结论
本研究建立了奶粉样品经提取、固相萃取柱净化,液相色谱-质谱联用仪分析的检测方法。该方法操作简单、快速,重现性好,定量准确,能满足奶粉样品中氯酸盐和高氯酸盐同时检测的要求,能为我国奶粉中氯酸盐和高氯酸盐残留情况的监控提供技术支撑。
猜你喜欢
氯碱工业(2022年1期)2022-07-02
口腔护理用品工业(2021年4期)2021-11-02
化工自动化及仪表(2021年3期)2021-06-04
食品工程(2020年4期)2021-01-20
中国油脂(2020年3期)2020-04-10
酿酒科技(2019年10期)2019-11-12
中成药(2018年6期)2018-07-11
中国氯碱(2017年4期)2017-05-04
中国粮油学报(2016年5期)2016-01-23
饮食科学(2015年7期)2015-11-23
