
高效液相色谱法测定双醋瑞因胶囊的有关物质
2019-09-13 03:28杨文凤张慧文
中国药业 2019年17期
杨文凤,石 颖,张慧文
(广东省广州市药品检验所,广东 广州 510160)
双醋瑞因(C19H12O8)属蒽醌类药物,化学名为4,5-二乙酰氧基 -9,10-二氢 -9,10-二氧代 -2蒽羧酸,主要活性代谢产物为大黄酸[1],是骨关节炎发病机制中参与重要炎性反应的白细胞介素1、白细胞介素6和肿瘤坏死因子-α的抑制剂[2-4],具有抗炎、镇痛作用,抑制软骨降解及破骨细胞性骨破坏[5-8]的作用,用于治疗退行性关节疾病(骨关节炎及相关疾病),可显著改善骨关节炎及相关疾病引起的疼痛和关节功能障碍等。为提高检验效率,参照进口药品注册标准,本试验中采用高效液相色谱(HPLC)法测定双醋瑞因胶囊有关物质[9-12],修订系统适用性试验要求[13-16],结果各杂质出峰顺序不因色谱柱及流动相比例的改变而改变,且杂质峰易分离,便于辨识。现报道如下。
1 仪器与试药
1.1 仪器
Waters E2695型高效液相色谱仪,含Empower工作站、Waters 2489型紫外检测器(美国 Waters公司);MT-5型电子天平(百万分之一)、XS105 DualRange型电子天平(十万分之一),均购于瑞士梅特勒-托利多公司。
1.2 试药
样品:双醋瑞因胶囊(某企业,批号分别为 A,B,C);峰识别对照品(某企业,批号为 371111,含量为100.5%,含双醋瑞因、单乙酰大黄酸Ⅰ、单乙酰大黄酸Ⅱ和大黄酸);大黄酸对照品(中国食品药品检定研究院,批号为 110757-200206,含量为 100.00%);醋酸(分析纯);二甲基甲酰胺(DMF,99.8%,Merck 公司,色谱纯)。
2 方法与结果
2.1 溶液制备
供试品溶液:避光操作,取装量差异项下粉末,研细,取细粉适量(约相当于双醋瑞因50 mg),精密称定,置50 mL容量瓶中,加DMF 40 mL,超声5 min,振摇使双醋瑞因溶解,加DMF稀释至刻度,摇匀,立即精密量取5 mL,置50 mL容量瓶中,用流动相乙腈-甲醇-醋酸溶液(20∶45∶35,V/V/V)稀释至刻度,摇匀,离心,取上清液,即得(临用新制)。
对照品溶液:取大黄酸对照品约10 mg,精密称定,置100 mL容量瓶中,加DMF 10 mL溶解,用流动相稀释至刻度,摇匀,精密量取1 mL,置100 mL容量瓶中,加流动相稀释至刻度,摇匀,即得。
对照溶液与灵敏度试验溶液:避光操作,精密量取供试品溶液1 mL,置100 mL容量瓶中,加流动相稀释至刻度,摇匀,作为对照溶液;精密量取对照溶液1 mL,置20 mL容量瓶中,加流动相稀释至刻度,摇匀,作为灵敏度试验溶液。
2.2 色谱条件与系统适用性试验
色谱柱:Kromasil C18柱(250 mm × 4.6 mm,5 μm);流动相:醋酸溶液(取水1 000 mL,用醋酸调pH至2.5)-甲醇 -乙腈(35∶45∶20,V/V/V);检测波长:254 nm;流速:1.0 mL /min;柱温:30 ℃ ;进样量:20 μL。理论板数按双醋瑞因峰计应不低于2 000。
取峰识别对照品适量,加DMF溶解并稀释成每1 mL约含1 mg的溶液,精密量取,加流动相稀释成每1 mL含 10 μg的溶液,量取20 μL,注入液相色谱仪,精密量取灵敏度试验溶液20 μL,注入液相色谱仪,双醋瑞因峰的信噪比大于10。再精密量取供试品溶液、对照溶液和对照品溶液各20 μL,分别注入液相色谱仪测定,记录色谱图至主成分峰保留时间的3倍。各组分的出峰顺序为双醋瑞因、单乙酰大黄酸Ⅰ、单乙酰大黄酸Ⅱ和大黄酸,相对保留时间分别为 1.0,1.4,1.8,2.2 min(图 1)。
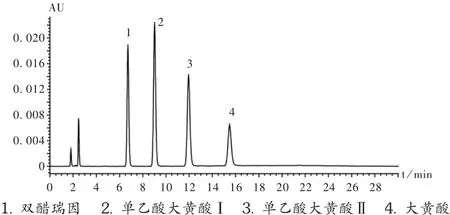
图1 系统适用性试验高效液相色谱图
2.3 专属性试验
取样品粉末适量(相当于含双醋瑞因125 mg),精密称定,置50 mL容量瓶中,加DMF溶解并稀释至刻度,摇匀,精密量取上述溶液5 mL,置50 mL容量瓶中,加流动相稀释至刻度,摇匀,作为未破坏样品贮备液。精密移取未破坏样品贮备液2.0 mL共4份,分别置10 mL容量瓶中,分别进行如下处理:加1 mol/L盐酸溶液2.0 mL,室温下放置 5 h后,加入 1 mol/L的氢氧化钠溶液适量,使之成中性,加流动相稀释至刻度,摇匀,过滤(酸破坏);加 1 mol/L 氢氧化钠溶液 2.0 mL,室温下放置30 min后,加入1 mol/L的盐酸溶液适量,使之成中性,加流动相稀释至刻度(碱破坏);加30%过氧化氢溶液2.0 mL,室温下放置5 h后,加流动相稀释至刻度(氧化破坏);放于光照强度为4 500 lx的实验箱中,室温下放置24h后,加流动相稀释至刻度(光照破坏)。另外,取双醋瑞因胶囊内容物(约相当于双醋瑞因125 mg),精密称定,放于密封的耐高温玻璃小瓶中,置80℃高温6 h,取出,放冷,转移至50 mL容量瓶中,加DMF溶解并稀释至刻度,摇匀(高温破坏)。精密量取上述条件处理溶液5 mL,分别置50 mL容量瓶中,加流动相稀释至刻度,按拟订色谱条件进样测定,记录色谱图。详见图2。
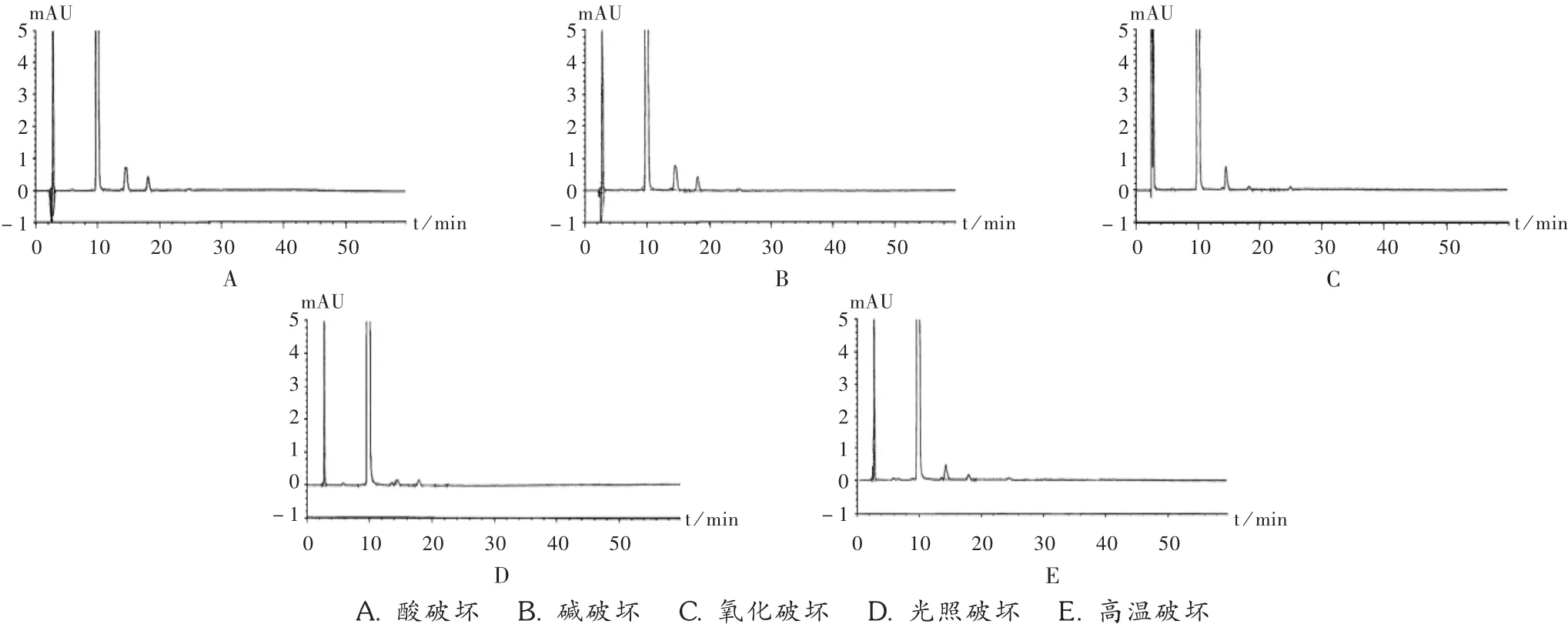
图2 专属性试验高效液相色谱图
可见,酸、碱、氧化、光照、高温破坏条件下降解产生的杂质峰不干扰主成分峰,主成分峰均一,色谱峰纯度大于990,峰纯度符合要求,无杂质峰与主成分一起洗脱。结果表明,双醋瑞因在光照、碱性条件下降解较明显,酸、高温、氧化条件下较稳定。样品记录色谱图至3倍主峰保留时间后无超过0.05%限度的降解杂质峰出现,在进行有关物质检查时样品记录色谱图至3倍主峰保留时间是合适的。同时也表明,本方法可稳定地检测样品所发生的降解变化,方法专属性强。
2.4 方法学考察
线性关系考察:取大黄酸对照品10.104 mg,精密称定,置100 mL容量瓶中,加入DMF 10 mL使溶解,加入流动相稀释至刻度,摇匀,精密量取1 mL,置50 mL容量瓶中,加流动相稀释至刻度,摇匀,作为大黄酸线性考察溶液(2.02 μg /mL);分别量取该溶液适量,置适宜容量瓶中,加流动相稀释成质量浓度分别为 1.20,1.00,0.60,0.20,0.08,0.04 μg /mL 的系列线性考察溶液。按拟订色谱条件测定,以质量浓度(C)为横坐标、峰面积(A)为纵坐标绘制标准曲线,得回归方程 A=83 963 C-787.71,r=0.999 9(n=7)。结果表明,大黄酸质量浓度在 0.04 ~2.02 μg/mL 范围内与峰面积线性关系良好。取双醋瑞因胶囊内容物0.262 6 g(平均装量每粒 0.246 31 g,含双醋瑞因 50 mg),精密称定,置50 mL容量瓶中,加DMF至刻度,摇匀;精密量取5 mL,置50 mL容量瓶中,加流动相稀释至刻度;再取1 mL,置50 mL容量瓶中,加流动相稀释至刻度,作为自身对照线性考察溶液(双醋瑞因质量浓度为 2.13 μg/mL),分别取该溶液适量,置容量瓶中,加流动相稀释制成质量浓度分别为 1.07,0.64,0.53,0.32,0.11,0.04,0.02 μg/mL的自身对照系列线性考察溶液。按拟订色谱条件测定,以质量浓度(C)为横坐标、峰面积(A)为纵坐标绘制标准曲线,得回归方程 A=6 110 430 C+342.29,r=1.000 0(n =8)。结果表明,双醋瑞因质量浓度在 0.02~2.13 μg/mL范围内与峰面积线性关系良好。
精密度试验:精密量取同一大黄酸对照品溶液,每次进样20 μL,重复进样6次,记录色谱图。结果的 RSD为 0.21%(n=6),表明仪器精密度良好。
检出限(LOD)和定量限(LOQ)确定:经试验,双醋瑞因及大黄酸的最低 LOD(S/N =3)分别为 0.006 μg/mL和 0.017 μg/mL,LOQ(S /N = 10)分别为 0.02 μg/mL和 0.058 μg /mL。
稳定性试验:取同一供试品溶液(批号C),分别于配置后的 0,2.5,6,17,19,33 h 时进样分析。结果随着时间的延长,主成分峰峰面积变化不明显,但单乙酰大黄酸Ⅰ和单乙酰大黄酸Ⅱ随着时间的延长峰面积逐渐增加,在33 h时含量分别由0.08%增加至0.16%、由0.05%增加至0.10%,但未见其他杂质峰增加,表明双醋瑞因在DMF中稳定性较差,应临用现配,立即用流动相稀释。
加样回收试验:取大黄酸对照品约5 mg,精密称定,置100 mL容量瓶中,加DMF溶解并定容至刻度,摇匀,作为贮备溶液。精密量取该贮备溶液1,2,3 mL(比例为50%,100%,150%),置加入相应空白辅料的100 mL容量瓶中,每个浓度样品溶液各制备3份,分别进样,记录峰面积,按外标法计算大黄酸含量,并计算回收率。结果见表1。
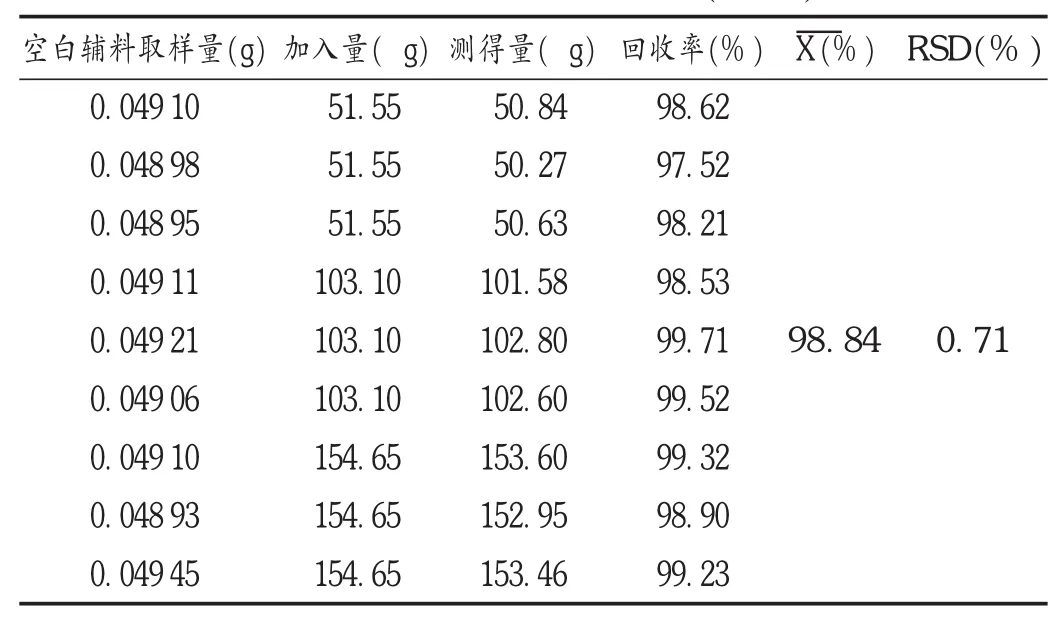
表1 大黄酸回收试验结果(n=9)
2.5 样品含量测定
按拟订色谱条件,对3批样品进行有关物质检查,用自身对照法计算各有关物质含量。结果见表2。
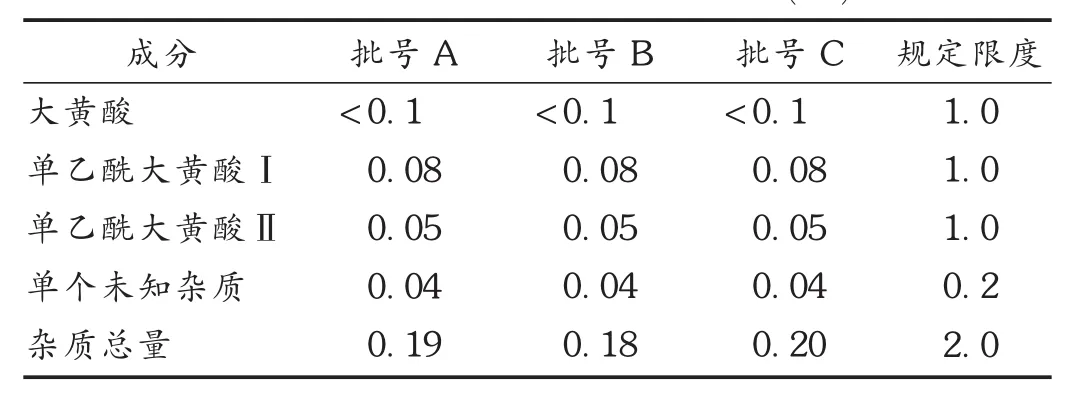
表2 3批样品有关物质检查结果(%)
3 讨论
分别考察了3个不同厂家品牌的色谱柱Kromasil C18柱 (250 mm ×4.6 mm,5 μm),Agilent HC-C18柱(250 mm ×4.6 mm,5 μm),GL ODS-3 柱(250 mm ×4.6 mm,5 μm),主峰与已知杂质分离度均大于 1.5,按双醋瑞因峰计理论板数均大于3 000,表明本试验中选定的色谱条件下,各组分峰形分离较好,适用于不同品牌C18柱,耐用性较好,满足检验需求。
在“药典在线”网上参照2010年版《印度药典》,采用与本方法相同的Kromasil C18柱(250 mm×4.6 mm,5 μm),流动相为 0.2%磷酸溶液 -乙腈(62∶38,V/V),检测波长为 254 nm,流速为 1.0 mL/min,柱温为 30℃,进样量为20 μL,测得单乙酰大黄酸Ⅰ、单乙酰大黄酸Ⅱ、大黄酸的分离度分别为 2.273,2.383,1.151,分离度较高,结果与本方法基本一致。
为考察色谱条件的微小变化对各有关物质测定结果的影响程度,本试验考察了流动相比例(醋酸水溶液 -甲醇 -乙腈 (30 ∶47.5 ∶22.5,V/V/V)即水相30% ,以及醋酸水溶液 -甲醇 -乙腈(40 ∶42.5 ∶17.5,V/V/V)即水相 40% 及 pH(±0.2)即 pH 2.3 及 pH 2.7的变化对测定结果的影响,有关物质测定结果差异不大(RSD<2.0%),均未检出其他最大单个未知杂质峰,表明本试验中选定的色谱条件下,各组分峰形分离较好。
本研究中色谱条件采用醋酸溶液(取水1 000 mL,用醋酸调节 pH 至 2.5)-甲醇 -乙腈(35∶45 ∶20,V/V/V)为流动相,各杂质的出峰顺序不易因色谱柱及流动相比例的改变而改变,在使用不同品牌C18柱时无差异,因此适用的色谱柱范围较广,避免因所用色谱柱柱效下降、分离度变差、已知杂质可能发生色谱峰重叠、未知杂质可能被误认为已知杂质而导致杂质误判的情况。同时,修订系统适用性试验要求,用含双醋瑞因、单乙酰大黄酸Ⅰ、单乙酰大黄酸Ⅱ和大黄酸的峰识别对照品为参比物,以相对保留时间作为系统适用性的要求和峰识别依据,方法更清晰、明了;由于该系统条件运行一针的时间短,能有效提高检验效率。
综上所述,本试验中建立的系统条件适用于双醋瑞因胶囊有关物质的测定,适用的色谱柱范围较广,各杂质峰易分离,试验效率高,结果准确可靠。
猜你喜欢
分子催化(2022年1期)2022-11-02
中国典型病例大全(2022年9期)2022-04-19
现代牧业(2021年4期)2021-04-14
中国应急管理科学(2021年4期)2021-04-13
核农学报(2020年7期)2020-07-01
中学生数理化·高一版(2020年9期)2020-01-02
学苑创造·A版(2019年9期)2019-11-07
中学化学(2019年4期)2019-08-06
考试周刊(2018年68期)2018-09-17
学苑创造·B版(2017年1期)2017-02-21
