
第一原理研究LuPO4中氧空位的性质
2019-09-12 06:28李金刘廷禹姚舒安付明雪鲁晓晓
无机材料学报 2019年8期
李金, 刘廷禹, 姚舒安, 付明雪, 鲁晓晓
第一原理研究LuPO4中氧空位的性质
李金, 刘廷禹, 姚舒安, 付明雪, 鲁晓晓
(上海理工大学 理学院, 上海 200093)
磷酸结构的晶体在掺杂二价阳离子后容易形成产生焦磷酸结构(P2O7)4–, 这种含有焦磷酸结构的氧化物材料十分适合做质子导体、燃料电池、气体传感器以及陶瓷膜等。本文利用第一性原理研究了LuPO4晶体中氧空位的结构性质, 结果显示当氧空位带二价正电时, 会引发氧空位周围原子奇特的畸变, 形成焦磷酸结构。为了解释这种结构畸变的机理, 本文利用过渡态搜索计算了结构变化过程中势能面的变化情况, 正一价氧空位形成焦磷酸 结构需要越过2.4 eV的势垒, 而正二价氧空位形成焦磷酸结构则不需要越过任何势垒, 因此很容易形成焦磷酸 结构。最后给出氧空位不同带电态的晶格结构、电子态密度以及电荷密度分布等基本物理性质, 氧空位处于正二价态结构下, 氧空位附近的P原子与O原子成键, 又由于O原子有较强的电负性, P的s轨道电子向O的p轨道转 移。P的s、p轨道在禁带中出现了与总态密度对应的缺陷能级, 结果表明带正二价氧空位的晶体性质发生了明显变化。
第一性原理; LuPO4; 氧空位; 过渡态搜索
磷酸结构的晶体在掺杂二价阳离子后会产生焦磷酸结构(P2O7)4–, 根据电荷补偿机制, 在替位的阳离子附近容易形成氧空位(焦磷酸结构)[1]。这种含有焦磷酸结构的氧化物材料十分适合做质子导体、燃料电池、气体传感器以及陶瓷膜等[1]。以LaPO4晶体为例, 在掺入正二价的Sr离子后, Sr2+替位La3+生成一个负电中心SrLa, 同时引入正电中心的氧空位(Vo)作为电荷补偿。LaPO4中的Vo是以焦磷酸结构的形式存在[2-3], 即两个邻近的PO4四面体共用一个氧原子。当材料处在潮湿环境中会引入水分子并与焦磷酸结合形成HPO4结构, 即(P2O7)4–+H2O(g)→2(HPO4)2–, 水分子的羟基进入氧空位, 余下的质子与另一个PO4四面体结合形成两个(HPO4)2–, (HPO4)2–的存在使得质子可以在LaPO4中传递。这种现象普遍存在于含有磷酸根(PO43–)的材料, 例如NdPO4、SmPO4、YPO4等[4-6]。
LuPO4材料有着稳定的化学性质等特点[7], 在掺杂Ce、Nd和Eu等元素后展现出良好的光学性能[8-11]。但至今为止很少有文献研究该材料的内部缺陷对其结构以及性能的影响。与YPO4和LaPO4有着类似的结构以及相似化学成分的LuPO4晶体, 本课题组研究发现当存在氧空位时同样也会形成焦磷酸结构, 这使得LuPO4也可能成为潜在的质子导体。最近, Li等[12]深入地研究了钙钛矿缺陷电荷对电子/空穴复合时间影响, 对缺陷电荷分配的研究有重要参考价值。
本课题组在研究不同电荷分布氧空位的结构稳定性及光谱性质时发现, 当氧空位处于正二价态时, 形成了焦磷酸结构[13], 实验也表明磷酸结构的晶体在氧空位附近很容易形成焦磷酸结构[1], 对其形成的机理目前仍不是很清楚, 本文针对该问题展开研究, 通过计算LuPO4晶体存在氧空位时的结构模型, 使用过渡态搜索(NEB)计算结构变化的最低能量路径以及能量的鞍点, 给出了带正二价的氧空位形成焦磷酸结构的机制。并讨论了氧空位处于不同价态下的晶格结构、电子态密度以及电荷密度分布。
1 计算方法和计算模型
选取96个原子LuPO4超原胞作为计算模型, 晶体的空间群为I41/amd[14-15]。如图1所示为四方锆石结构的LuPO4晶体, P原子被四个氧原子包围构成正四面体结构, Lu原子被八个氧原子包围形成十二面体结构。使用VASP软件[16-17]对LuPO4晶体进行模拟计算, 选取平面波赝势结合广义梯度近似法GGA-PBE处理交换相关能[18-20], K网格点选取为3×3×3, 截断能选取为450 eV, 以上参数均经过收敛测试确定。LuPO4晶体中的氧原子只有一种对称格位, 氧空位的模型是在超原胞中心附近挖去一个氧原子, 并对氧空位在0、+1、+2价态下做结构弛豫以及静态计算。不同价态的氧空位优化后, 结构区别最大的是氧空位最近邻的一个P原子。过渡态搜索方法为: 完整晶体挖去一个氧原子做结构优化的结构作为Nudged Elastic Band (NEB)计算的初态, 而含带两个正电荷的氧空位晶体做结构优化的结果(含焦磷酸结构(P2O7)4–结构)作为末态, 在初态以及末态中线性插入四个点作为过渡态, 并让氧空位分别带+1价和+2价做NEB计算[21]。比较+1价与+2价的计算结果, 得出结构变化的机理。

图1 四方锆石结构的LuPO4超原胞
2 结果与分析
2.1 P原子的位移
通过对含有氧空位的LuPO4晶格做结构弛豫, 不同价态下的结构驰豫结果如图2所示。P原子被四个氧原子包围构成正四面体结构, 当除去在P原子附近一个氧原子形成氧空位后, P原子的位置会随着价态的变化而发生改变, 向着与氧空位相反的方向移动。当氧空位处于正二价态时, P原子会穿过三个氧原子(O(5)、O(6)和O(7))构成的势垒面到达这个面的另一侧, 并重新键连O原子形成一个新的正四面体结构。如图2所示P(2)与O(4)键连并与左侧的一个完整的(PO4)3–四面体结合形成一个新的焦磷酸结构[P2O7]4–。
表1表示P原子与附近原子间的键长, 可以看出在完整结构下P原子与近邻的O原子间距近似为0.155 nm, P原子处在正四面体的中心位置。氧空位处于电中性时, 与VO最近邻的原子将向着空位方向移动。从表格中也可以看出氧空位处于电中性下P(2)与P(1)、O(4)以及近邻的三个氧原子的距离有所增大, 意味着P原子向着空位方向移动。通过P(2)与近邻的三个氧原子(O(5)、O(6)、O(7))的距离可以看出随着带电数的增加P是向远离VO的方向移动。由于正电荷局域在氧空位附近, 又由于正五价的P原子与正二价的氧空位有较强的库仑排斥作用使得P原子附近结构发生了急剧变化形成了焦磷酸结构(P2O7)4–。通过观察P(1)与O(4)的键长发现随着P(2)的靠近O(4)也在向着与P原子成键的方向移动, 成键后的O(4)与P(1)、P(2)的键长分别为0.162和0.168 nm, 相较于完整结构下P-O键均有所拉长。
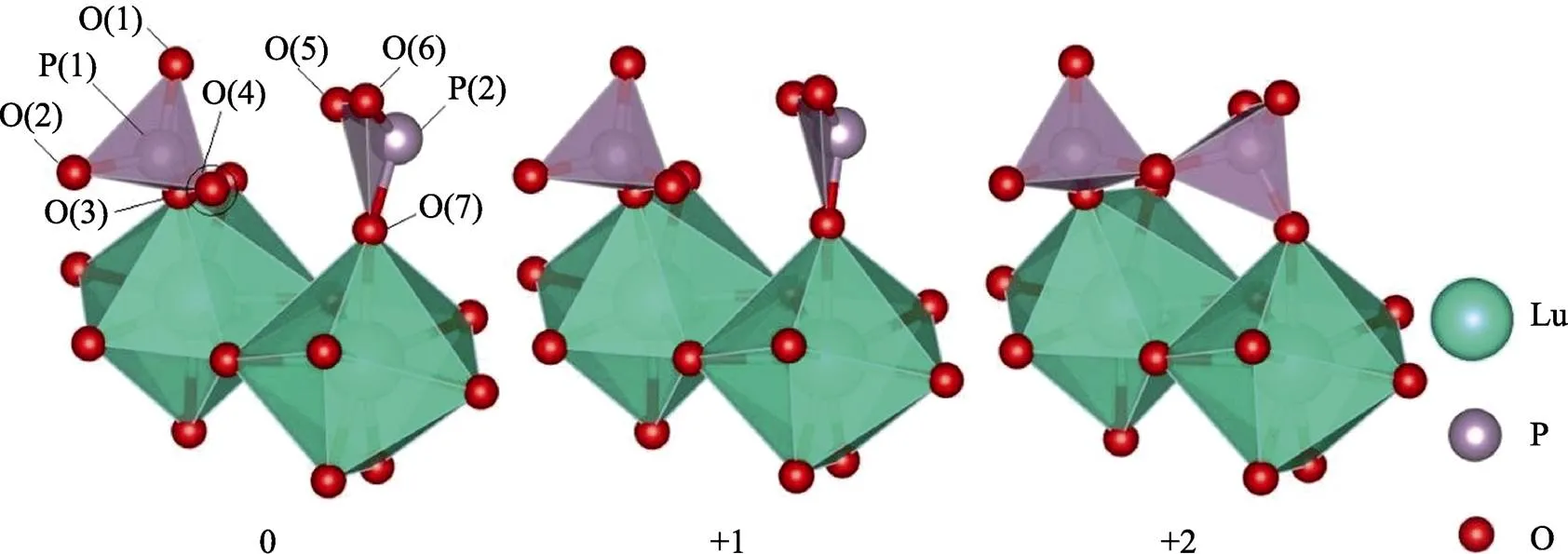
图2 含Vo的LuPO4晶格在不同价态下的结构
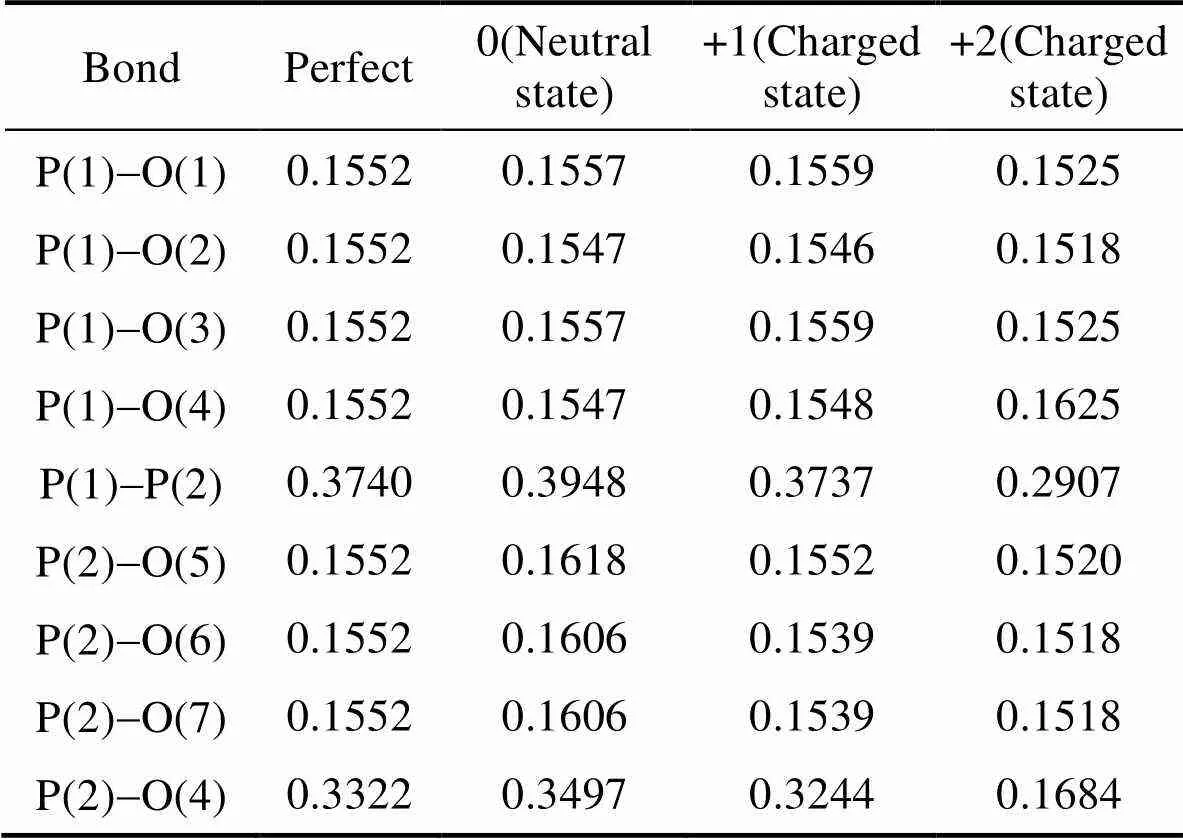
表1 完整超原胞和含不同价态下氧空位晶体的结构参数(nm)
2.2 过渡态搜索
过渡态搜索是一种已知反应物和生成物而寻找能量的最小路径以及鞍点的方法[21], 使用完整晶体挖去一个氧原子做结构优化的结构作为NEB计算的初态, 而带两个正电荷的氧空位晶体作结构优化结果作为末态, 采用线性插值作为插入点的算法并插入四个过程的中间态。当氧空位带正二价时, 相应结构的能量从初态到末态呈单调递减趋势, 如图3所示。这意味结构变化的过程中没有势垒, 初态结构的能量较高, 晶格会随着驰豫过程自发地向末态结构变化。含正一价的氧空位晶体过渡态搜索结果用高斯曲线拟合, 结果如图4所示。横坐标表示原子移动的距离, 纵坐标表示相对于初态能量每个结构下的势能。峰值能量大约为2.4 eV, 可以看出结构变化中需要越过的势垒较高, 对于正一价氧空位要形成焦磷酸结构从能量角度考虑比较困难。而正二价氧空位形成焦磷酸结构则不需要越过任何势垒,很容易形成焦磷酸结构。图4中只显示了五个点是因为末态的能量急剧降低为-4.45 eV无法拟合成高斯函数, 这可能因为形成了焦磷酸结构, 能量急剧降低后在这个结构下达到稳定, 这也从侧面印证了在氧空位存在的情况下焦磷酸结构比较稳定。
图5表示LuPO4晶格在氧空位处于不同价态下的电荷密度分布, 可以看出Lu原子周围的电荷呈圆形分布, 说明Lu主要为离子性。O原子周围电荷分布存在明显方向性, O原子有较强的电负性, O原子与P原子之间以共价键为主。图中可以看出随着氧空位带正电数的增加, P原子与O原子向着成键的方向移动, 最终一个氧原子将会与两个P原子成键并形成焦磷酸结构, 这个过程中P原子从有一个悬挂键到与O4形成共价键, 需要与O4共享电子, 也就是O4将可能失去部分电子给P原子, 说明O4可能发生了氧化反应。
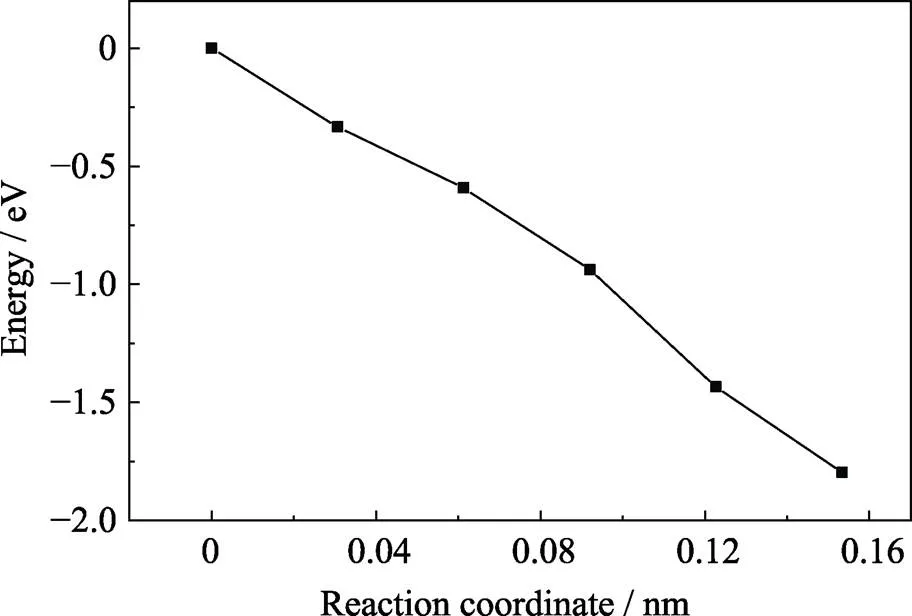
图3 过渡态搜索计算得到的能量路径(Vo处于正二价态)
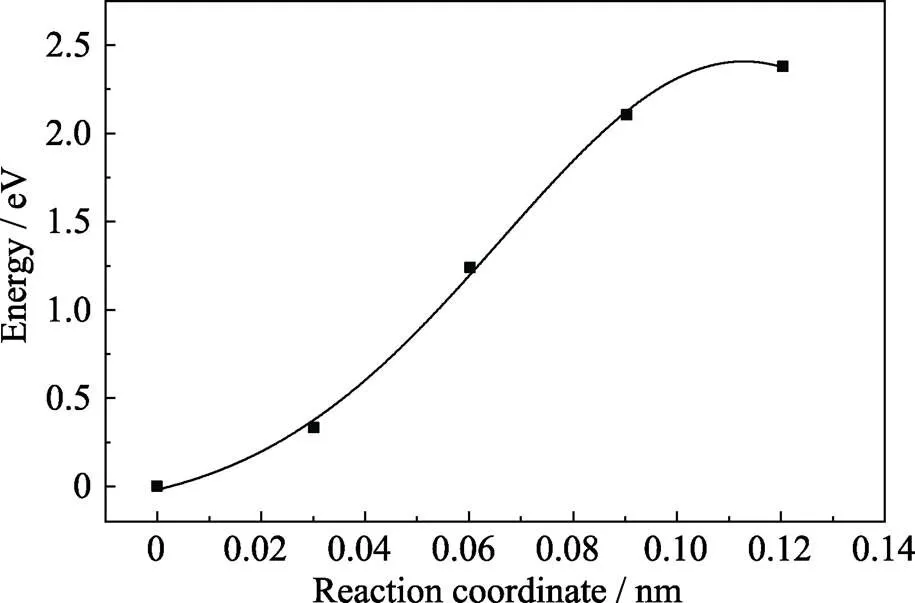
图4 过渡态搜索计算的最低能量路径(Vo处于正一价态), 峰值大约2.4 eV
2.3 电子态密度
含不同价态氧空位的LuPO4晶体态密度以及完整结构的总态密度如图6所示, 从完整结构的态密度图可以看出LuPO4的带隙5.8 eV相比实验带隙值8.6 eV[22]偏小, 这是由于密度泛函理论对交换相关项处理不够从而低估了材料的带隙值[8], 但并不影响对态密度的定性分析。完整晶体价带顶主要由Lu的4f轨道和O的2p轨道构成, 导带底主要由Lu的5d轨道和少量P的3s、3p轨道以及O的2p轨道构成。深能级分别是由Lu的p轨道和O的s、p轨道构成以及少量P的s、p轨道构成价带顶的尖峰大约在-3.5 eV是由Lu的4f轨道构成, 峰值在-24 eV的能量来自于Lu的5p态,-20.5 eV来自O的2s态,-17.5 eV主要是由O的2s态构成。靠近价带顶的两个能量区间-4.5~-6 eV、-7~8.5 eV分别来自O的2p轨道与P的3p轨道以及O的2s、2p轨道与P的3s轨道的杂化。
通过分析图6发现氧空位处于电中性时在禁带中出现一个由O的2p轨道构成的缺陷能级, 距离价带顶大约1 eV。当氧空位带一价正电时在价带顶和导带底附近分别各出现一个缺陷能级, 其中距价带顶0.7 eV的缺陷能级同样是由O的2p态构成, 距离导带底0.4 eV的缺陷能级是由Lu的5d轨道构成, 推测可能是由于配位场的改变导致Lu的能级分裂, 因而在禁带之中产生了缺陷能级。带隙中存在缺陷能级会影响材料的光学性能, 这类靠近带边的缺陷能级可能会成为一个浅陷阱, 抓住电子或空穴, 当LuPO4晶体掺入发光中心作为闪烁晶体时, 这类浅陷阱可能会延长晶体的发光时间, 产生慢发光成分从而影响材料的光学性能[23]。当氧空位带正二价电时Lu的5d态构成的缺陷能级距离导带底大约0.7 eV,这是由于氧空位附近的晶格畸变更加明显, 导致附近Lu离子的配位场进一步改变, 使得Lu离子的外层电子轨道分裂更加明显, 从而引入的缺陷能级距离导带底更远。当吸收光子能量时离化的施主可能会和价带之间产生激发跃迁, 而这类靠近导带底的缺陷能级参与的吸收光能量将非常靠近带边吸收的能量。在LuPO4:Nd晶体中发现存在一个由缺陷引起的紫外吸收[24], 吸收对应的能量非常靠近带边吸收, 这种吸收很可能与氧空位的存在有关[13]。
本文进一步研究了氧空位附近变化最明显的O(4)原子和P(2)两个原子的态密度分布, O(4)原子p轨道的分波态密度如图7所示。正二价态下的态密度变化最明显, 这是由于形成了[P2O7]4–的结构使得O(4)的对称性破坏能级发生分裂, 导致O(4)原子的态密度上出现更多特征明显的尖峰。其中氧空位处于正二价态下出现3个明显的尖峰, 峰值分别位于-1.2、-4.3和-6 eV, 而价带顶附近其他峰值的态密度有所减弱, 这说明在氧空位处于正二价态时, 这三个峰的局域性较强。从图中还可以看出带隙中出现的缺陷能级与总态密度的缺陷能级对应, 是由于Vo的存在使邻近的O(4)原子配位场发生改变产生能级分裂, 因而在禁带中产生了由O(4)的p轨道构成的缺陷能级。P(2)原子s、p轨道的分态密度如图8和图9所示。与O(4)的p轨道不同P(2)的p轨道变化并不明显如图9所示, 态密度的变化主要来自s轨道。图8可以看出在氧空位处于正一价态下, P(2)原子s轨道主要局域在-4.3 eV附近, 但当氧空位处于正二价态时该峰值消失并且态密度有所下降。通过比对O(4)在正二价的s轨道发现在-4.3 eV附近出现了明显的峰值, 推测可能是由于在氧空位处于正二价态结构下P(2)原子与O(4)原子成键。P(2)的s、p轨道在禁带中出现了与总态密度对应的缺陷能级, 说明氧空位对P(2)原子的配位场存在明显影响, 使得原子的能级产生分裂并引入到禁带中。
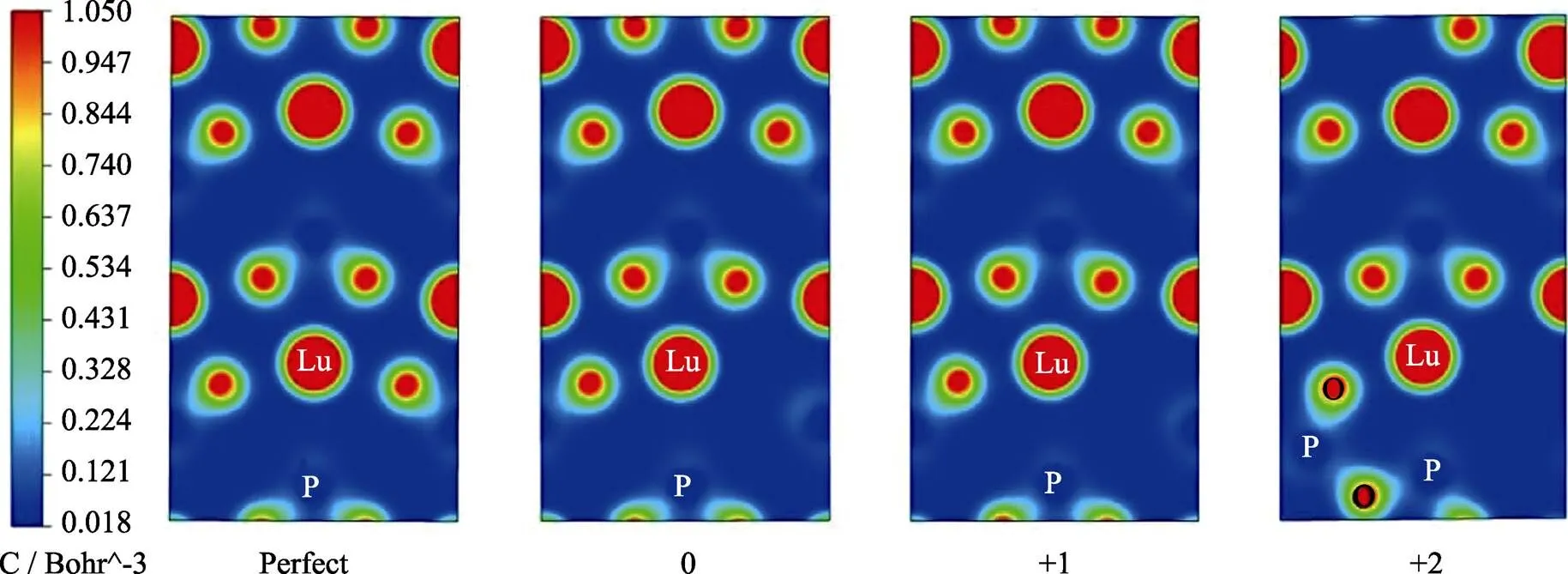
图5 完整结构以及含缺陷晶格的电荷密度分布图
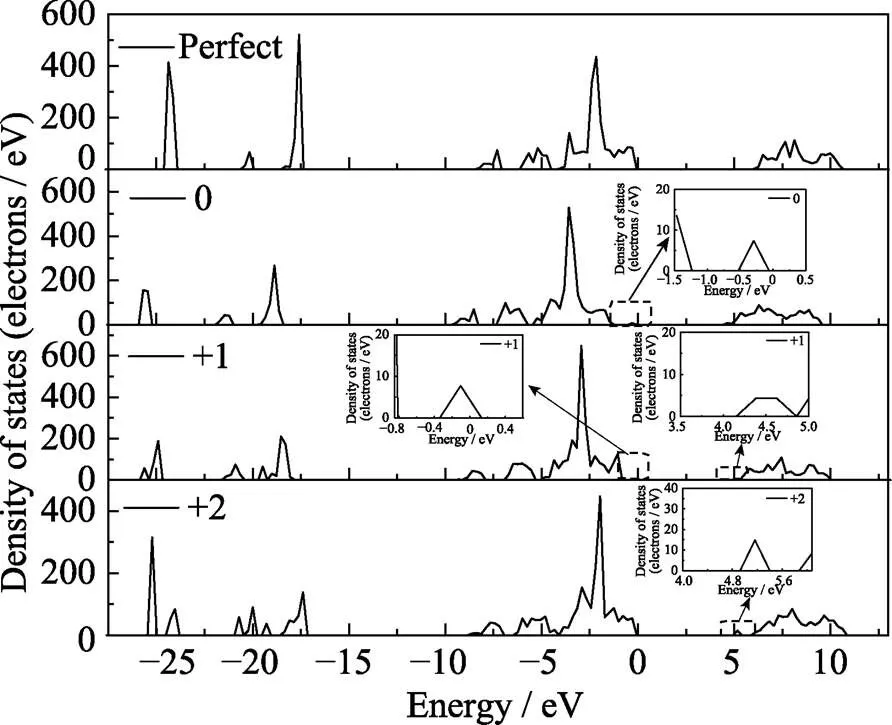
图6 不同结构下总态密度图
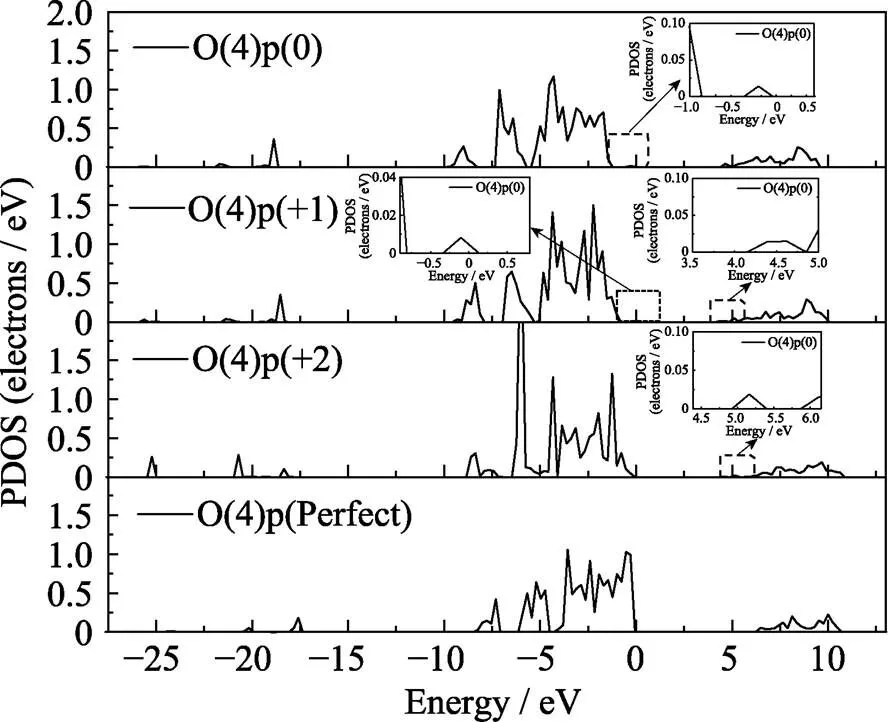
图7 不同结构下O(4)原子p轨道的分波态密度图
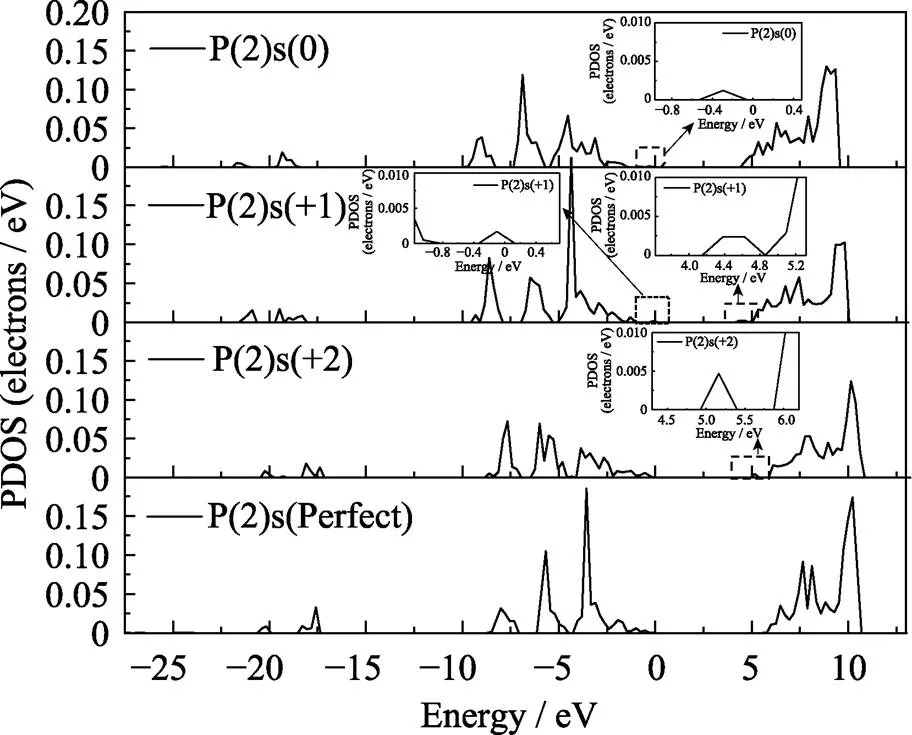
图8 不同结构下P(2)原子s轨道的分波态密度图
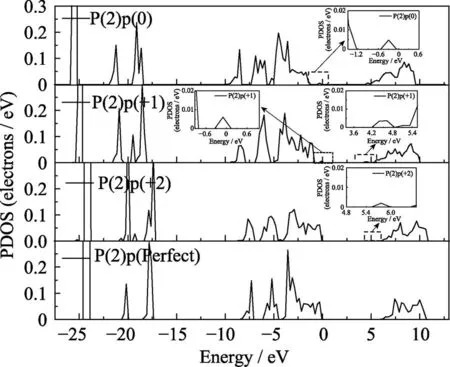
图9 不同结构下P(2)原子p轨道的分波态密度图
3 结论
本文通过密度泛函理论下的广义梯度近似结合平面波雁势方法对含有Vo的LuPO4的晶体进行了研究。计算了不同价态的Vo在LuPO4中的结构, 当Vo带正二价电时, P原子会越过由三个氧原子构成的势垒面, 并重新键连一个O原子形成焦磷酸结构(P2O7)4–。使用过渡态搜索计算结构变化过程中势能面的鞍点, 从势能角度验证了带正二价氧空位周围晶格发生奇特的畸变的机理。并讨论了氧空位处于不同价态时对应结构的电荷密度分布以及电子态密度。带正二价氧空位的晶体性质发生了明显变化。LuPO4:Nd晶体中靠近带边吸收很可能与氧空位的存在有关。
[1] PHADKEP S, NINO J C, ISLAM M S. Structural and defectproperties ofthe LaPO4and LaP5O14-basedproton conductors., 2012, 22(48): 25388–25394.
[2] SOLOMON J, ADELSTEIN N, ASTA M,. Charge-compensating pyrophosphate defect structures in Sr-doped LaPO4., 2012, 45(1): 117–120.
[3] AMEZAWZ K, MAEKAWA H, TOMII Y,Protonic conduction and defect structures in Sr-doped LaPO4., 2001, 145(1–4): 233–240.
[4] KITAMURA N, AMEZAWA K, TOMII Y,High temperature protonic conduction in Sr-doped NdPO4., 2002, 49(10): 856–860.
[5] KITAMURA N,AMEZAWA K,TOMII Y,Protonic conduction in rare earth orthophosphates with the monazite structure., 2003, 162: 161–165.
[6] AMEZAWA K, TOMII Y, YAMAMOTO N. High temperature protonic conduction in Ca-doped YPO4., 2003, 162: 175–180.
[7] RAPAPORT A, DAVID V, BASS M,Optical spectroscopy of erbium-doped lutetium orthophosphate., 1999, 85(1/2/3): 155–161.
[8] SUN C, LI X, WANG H,Crystallization-dependent luminescence properties of Ce: LuPO4., 2016, 55(6): 2969–2976.
[9] LEMPICKI A, BERMAN E, WOJTOWICZ A J,Cerium- doped orthophosphates: new promising scintillators., 1993, 40(4): 384–387.
[10] MAKHOV V N, KIRIKOVA N Y, KIRM M,Luminescence properties of YPO4: Nd3+: a promising VUV scintillator material., 2002, 486(1/2): 437–442.
[11] VISTOVSKYY V, MALYI T, VAS’KIV A,Luminescent properties of LuPO4-Pr and LuPO4-Eu nanoparticles., 2016, 179: 527–532.
[12] LI WEI, SUN YI-YANG, LI LIN-QIN. Control of charge recombination in perovskites by oxidation state of halide vacancy., 2018, 140(46): 15753–15763.
[13] LI JING, LIU TINGYU, FU MINGYUE,Optical properties simulating calculation of the F or F+center in LuPO4crystal., 2018, 47(7): 1–7.
[14] NIPKO J C, LOONG C K, LOEWENHAUPT M,Lattice dynamics of xenotime: the phonon dispersion relations and density of states of LuPO4., 1997, 56(18): 11584–11592.
[15] NIPKO J C, LOONG C K, LOEWENHAUPT M,. Lattice Dynamics of LuPO4., 1997, 250(1/2): 573–576.
[16] KRESSE G, FURTHMULLER J. Efficient iterative schemes fortotal-energy calculations using a plane-wave basis set., 1996, 54(16): 11169–11186.
[17] KRESSE G, FURTHMULLER J. Efficiency oftotal energy calculations for metals and semiconductors using a plane- wave basis set., 1996, 6(1): 15–50.
[18] PERKEW J P, BURKE K, ERNZERHOF M. Generalized gradient approximation made simple., 1996, 77(18): 3865–3868.
[19] KRESSE G, JOUBERT D. From ultrasoft pseudopotentials to the projector augmented-wave method., 1999, 59(3): 1758–1775.
[20] BLOCHL P E. Projector augmented-wave method., 1994, 50(24): 17953–17979.
[21] HENKELMAN G, JUAO NSSON H. Improved tangent estimate in the nudged elastic band method for finding minimum energy paths and saddle points., 2000, 113(22): 9978–9985.
[22] MIKHAILIN V V, SPASSKY D A, KOLOBANOV V N,Luminescence study of the LuBO3and LuPO4doped with RE3+., 2010, 45(3–6): 307–310.
[23] WISNIEWSKI D, TAVERNIEER S, WOJTOWICZ A J,LuPO4:Nd and YPO4:Nd—new promising VUV scintillation materials., 2002, 486(1/2): 239–243.
[24] WISNIEWSKI D, TAVERNIER S DORENBOS P,VUV Scintillation of LuPO4:Nd and YPO4:Nd., 2002, 49(3): 937–940.
First Principles Study on the Property of O Vacancy in LuPO4Crystal
LI Jin, LIU Ting-Yu, YAO Shu-An, FU Ming-Xue, LU Xiao-Xiao
(College of Science, University of Shanghai for Science and Technology, Shanghai 200093, China)
Phosphoric acid crystal doped with bivalent cation is easy to form pyrophosphate structure (P2O7)4–. This kind of oxide material containing pyrophosphate structure is very suitable for proton conductor, fuel cell, gas sensor, and ceramic film, etc. In this research we applied first principles to study the structural behaviour of oxygen vacancy in LuPO4crystal. The results showed that oxygen vacancies with two positive charges distort structure largely and form pyrophosphate structure. In order to verify the feasibility of this structure transition, the nudged elastic band method is used to find the highest saddle point of potential energy. The calculated results show that transition state energies of oxygen vacancy with +1 and +2 charge forming pyrophosphate structure are about 2.4 and 0 eV, respectively. So pyrophosphate structure easily forms for oxygen vacancy with +2 charge. Finally, the lattice structure, density of states and charge density distribution are obtained. P atom and O atom around the oxygen vacancy with +2 charge can form chemical bond. The electron on the P s will shift to O p orbit for oxygen with strong electronegative, which introduces the defect level in forbidden band. It is indicated that the property of the crystal changes drastically for the existence of the oxygen vacancy with two positive charges.
first principle; LuPO4; oxygen vacancy; nudged elastic band method
O77
A
1000-324X(2019)08-0879-06
10.15541/jim20180431
2018-09-12;
2019-01-27
国家自然科学基金(61675132) National Natural Science Foundation of China (61675132)
李金(1994–), 男, 硕士研究生. E-mail: 995084819@qq.com
刘廷禹, 教授. E-mail: Liutyyxj@163.com
猜你喜欢
当代党员(2022年9期)2022-05-20
华人时刊(2021年23期)2021-03-08
复旦学报(医学版)(2020年3期)2020-06-18
伴侣(2020年4期)2020-04-27
校园英语·月末(2019年11期)2019-09-10
保健与生活(2019年3期)2019-08-01
作文中学版(2018年1期)2018-11-28
发明与创新·大科技(2018年5期)2018-09-19
中学生数理化·高三版(2017年1期)2017-04-20
读者欣赏(2014年6期)2014-07-03
