
TLC-UPLC-MS/MS 法检测网红保健品中非法添加的8 种化学药物
2019-09-06 07:53:22曹晓琴方振峰周国勇陈中强
食品科学 2019年16期
曹晓琴,方振峰,张 涛,周国勇,陈中强,施 璐*
(江汉大学医学院,湖北 武汉 430056)
目前,一些不法厂商为谋取经济利益,盲目追求疗效,在网络销售的保健食品中非法添加化学药物,变“禁药”成“网红药”,严重危害人民群众的生命健康安全[1]。其中,酚酞和西布曲明是网红减肥药中常见的添加物[2-3],一些商家还将麻黄碱和咖啡因联合作为减肥的辅助药物添加[4-5];西地那非是网红补肾壮阳类保健品中常见的添加物[6];洛伐他汀和辛伐他汀是网红降脂类保健品中常见的添加物[7];苯乙双胍是网红降糖类保健品中常见的添加物[8]。同时,日常检验过程中,还发现一些减肥类、降血脂类和润肠通便类保健食品在功能宣称和非法添加的药物类型上往往具有交叉,既能迅速达到所宣称的功效,又规避了非法添加化学药物的检测[9-10],而网售药品和保健品作为一种新生事物,监管体系还不健全,因此急需针对网红保健品建立全面有效的检测方法,打击网上“多种类”、隐蔽性强的非法添加行为。
目前,保健品中非法添加西药的常用检测方法有薄层色谱(thin-layer chromatography,TLC)法[11]、高效液相色谱(high performance liquid chromatography,HPLC)法[12-14]、超高效液相色谱(ultra performance liquid chromatography,UPLC)法[15-16]、显微傅里叶变换红外光谱(micro fourier transform infrared spectroscopy,Micro FTIR)法[17]、表面增强拉曼光谱(surface-enhanced raman spectroscopy,SERS)法[18]、高效液相色谱-串联质谱联用(high performance liquid chromatography-tandem mass spectrometry,HPLC-MS/MS)法[19-21]等。TLC和HPLC由于仪器性能限制问题,灵敏度低,容易造成假阳性的结果[11]。UPLC比传统的液相色谱具有更高的分辨率和更快的分析速度,但是无法做到定量确证[15],HPLC-MS/MS法是目前监测非法添加的有力工具,但是,文献调研发现,大部分监测的对象仅集中在各自的小类里[22-23],而非法添加的网红保健品为达到宣称的功能疗效,在非法添加的药物类型上往往具有交叉,大大增加了检测的难度。因此,本研究依据相关报道[24-26],选取了8 种具有减肥、降脂、通便功能的代表性药物,同时也是从网红商家处方便易得的一些代表性药物,采用UPLC-MS/MS法进行检测。在前处理过程中,大部分文献采取用甲醇或乙腈直接提取的方式进行前处理[15,21],对一些复杂的基质并没有进行针对性的处理,只是采用过滤膜去除杂质干扰[24-26]。也有研究为避免基质干扰,采用固相萃取法(solid-phase extraction,SPE)进行前处理,但是操作过程复杂,成本较高[27-29]。
本研究将TLC法应用到样品前处理中,不仅避免了常规有机试剂提取方法容易造成干扰大、漏检、假阴性的结果[9-11],也摒弃了文献采用SPE法前处理成本高、较为复杂且不适合大量筛查的缺点[28-29],同时可以做到操作简单,结果准确。针对网红保健食品中非法添加化学成分检测方法的缺失和标准落后的情况,本实验将TLC法应用到样品前处理过程中,再联用高灵敏度、高专一性的UPLC-MS/MS法,拟建立同时检测网红保健品中非法添加8 种不同类别的药物(西布曲明、洛伐他汀、辛伐他汀、麻黄碱、咖啡因、酚酞、苯乙双胍和西地那非)的TLC-UPLC-MS/MS方法。并采用该方法对市售45 种网红保健食品进行检测,以期为打击非法添加、药品监管及规范网络市场提供技术支撑。
1 材料与方法
1.1 材料与试剂
盐酸西布曲明(纯度99.7%)、洛伐他汀(纯度99.4%)、辛伐他汀(纯度99.0%)、盐酸麻黄碱(纯度99.7%)、咖啡因(纯度99.9%)、酚酞(纯度99.9%)、盐酸苯乙双胍(纯度99.7%) 中国食品药品检定研究院;枸橼酸西地那非(纯度99.5%) 美国Sigma-Aldrich公司;甲醇、乙腈、甲酸铵、甲酸、乙酸铵、乙酸(均为色谱纯) 美国Fisher Chemicals公司;去离子水为Millipore公司超纯水器制备;氯仿、氨水、氢氧化钠、盐酸(均为分析纯)、羧甲基纤维素钠(批号:20161220) 国药集团化学试剂有限公司;GF254薄层硅胶板(200 mm×100 mm) 青岛海浪硅胶干燥剂厂。
45 批保健食品样品均来自网购。
1.2 仪器与设备
API 4500三重四极杆MS/MS 美国Applied Biosystems公司;UPLC 日本Shimadzu公司;Milli-Q超纯水系统 美国Millipore公司;离心机 湖南湘立科学有限公司;Kinetex 100 RP-18色谱柱 美国Phenomenex公司。
1.3 方法
1.3.1 色谱条件
色谱柱(100 mm×2.1 mm,2.6 μm),柱温:35 ℃;进样量:10 µL;流速:0.2 mL/min。流动相梯度见表1。
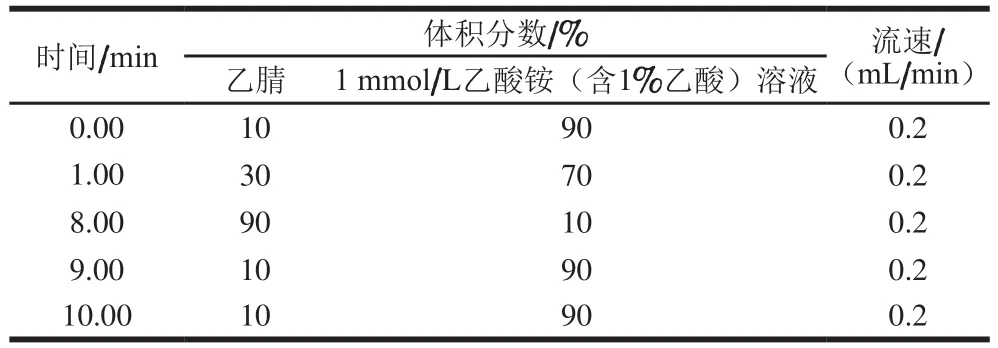
表1 UPLC梯度洗脱程序Table 1 Mobile phase gradient program of UPLC
1.3.2 质谱条件
结合文献[19-21]报道,本研究分别采用乙腈-水、5 mmol/L乙酸铵-乙腈、0.2 mmol/L乙酸-乙腈的溶剂体系(50∶50,V/V)配制10 µg/L的标准溶液,依次进行质谱条件的选择。采用参数分别为:离子源:电喷雾电离(electrospray ionization,ESI)源;扫描方式:正离子模式进行扫描;气帘气15 psi;雾化气(Gas1):55 psi;辅助气(Gas2):50 psi;离子喷雾电压:5 500 V;离子源温度:550 ℃;在多重反应监测(multiple reaction monitoring,MRM)状态下,通过优化离子对的碰撞能量和去簇电压值,得到各目标化合物的质谱参数见表2。
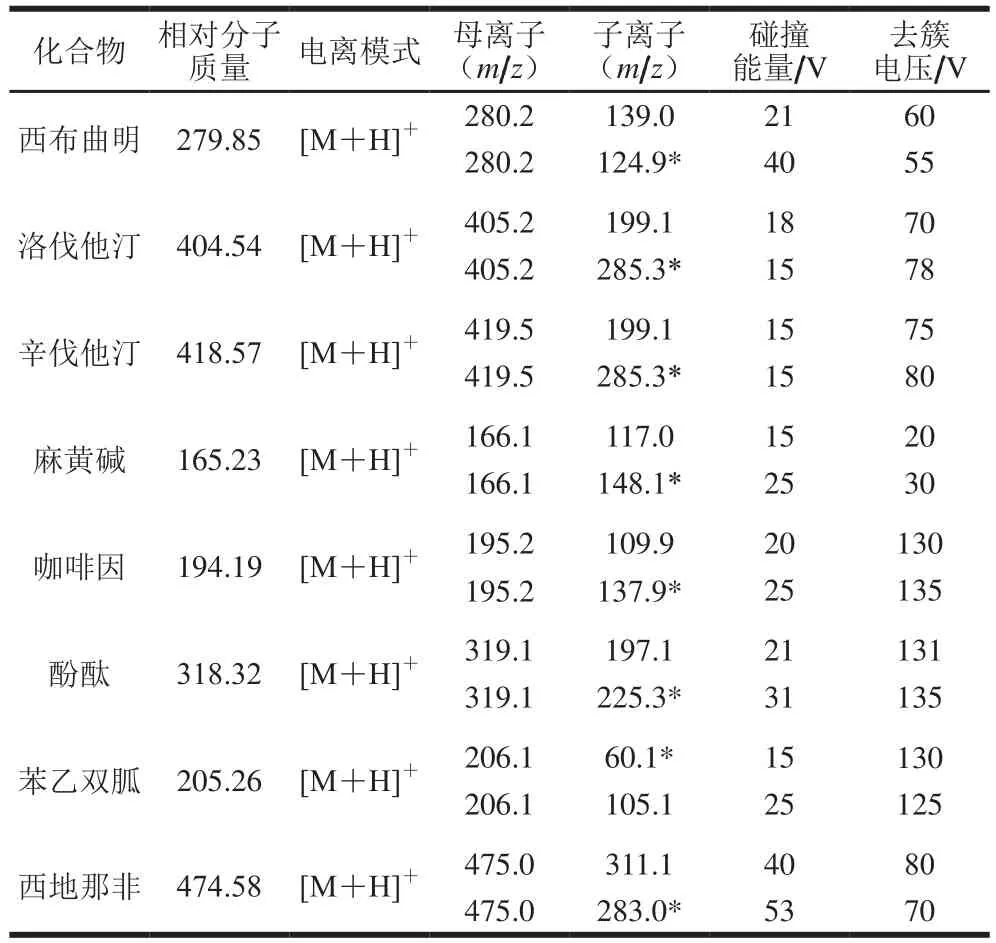
表2 8 种药物在MRM模式下的母离子和子离子信息Table 2 Precursor/daughter ions information of the 8 drugs in the MRM mode
1.3.3 对照品溶液的制备
标准对照品贮备液的配制:精密称取各对照品10.0 mg,用10 mL甲醇溶解、定容,分别制成质量浓度为1.0 mg/mL标准贮备液,-20 ℃避光保存。
混合对照品标准工作液的配制:精密量取标准贮备液各0.1 mL,加入空白样品提取液中,定容到10 mL,制成质量浓度为10.0 µg/mL的混合标准工作液,4 ℃避光保存。
1.3.4 供试品的制备
片剂:随机取同一批号的供试品20 片,研细为试样;硬胶囊:随机取同一批号的供试品20 粒,倾出所有内容物,研细为试样;软胶囊:随机取同一批号的供试品20 粒,将内容物全部挤到同一离心管中,充分混匀后,作为试样;口服溶液剂:随机取同一批号的供试品10 支(瓶),取等量体积样品到同一容器中混匀,作为试样;其他固体类样品(饼干、糖果、粉末、茶剂):随机取同一批号的供试品10 份,研细或粉碎为试样。
称取上述处理好的试样约1.0 g,置于5 mL离心管中,加入2 mL甲醇,超声提取(功率143 W;频率40 kHz)15 min后,4 000 r/min离心5 min,取上清液作为供试品溶液。
所有采集的样品均采用建立的检测方法进行空白基质的筛选,测定结果为无任何干扰峰的基质作为空白基质,用于样品添加实验。
1.3.5 前处理条件
本研究采用直接提取法、SPE法和TLC法分别进行实验。
直接提取法采用文献[21]方法,称取1.3.4节处理好的试样约1.0 g,置于5 mL 离心管中,加入2 mL甲醇,超声提取(功率143 W;频率40 kHz)15 min后,4 000 r/min离心5 min,取上清液,过0.22 μm滤膜上机检测。
SPE法参考文献[28-29]方法,采用Waters Oasis HLB SPE小柱1 mL/30 mg,依次加入甲醇3 mL和水3 mL活化后,经1.3.4节制备好的供试品溶液上样,再用甲醇-水(50∶50,V/V)3 mL淋洗,抽干后,用甲醇3 mL洗脱,收集洗脱液。氮气吹干,1 mL流动相复溶,过0.22 μm 滤膜,续滤液作为供试品溶液。
TLC法在文献[17-18]基础上进行适当调整,以硅胶GF254薄层板为固定相,以氯仿-甲醇-氨水(11∶1∶0.1,V/V)为展开剂,取制备好的供试品溶液点于薄层板上,点样量为0.5 mL,展开后取出晾干,在紫外灯254 nm下检视定位。将显色的薄层挖下来放入1.5 mL EP管中,将薄层粉末连同EP管一同烘干后,用0.5 mL的甲醇溶解,超声 5 min、高速离心3~5 min,取上清液,过滤膜上机检测。
1.3.6 定量测定
空白样品提取液中加入1.3.3节制备的对照品溶液制成混合标准溶液,在1.3.1节和1.3.2节的色谱和质谱条件下进样分析,以各目标物质的响应信号强度y为纵坐标,对应药物的浓度为横坐标绘制标准工作曲线。样品溶液中各待测药物响应值均应在测定的线性范围内。
1.4 统计学分析
在优化条件和实际样品测定时,平行实验重复操作6 次,并采用SPSS 13.0(Chicago, IL, USA)统计软件分析。基质效应通过t检验考察[30]。
2 结果与分析
2.1 质谱条件的选择
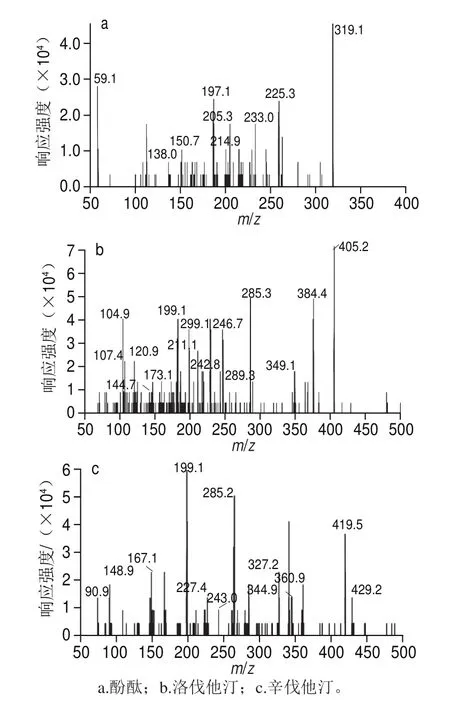
图1 部分药物的二级质谱图Fig. 1 Mass spectra of selected drugs
质谱条件选择时,ESI的正负离子模式均被评估,结果发现,待测物均可选择[M+H]+离子作为母离子。这与许立等[9]报道一致。而洛伐他汀和辛伐他汀很容易加和成[M+Na]+模式,而且响应很高,但其二级碎片离子稳定性较差,而响应较弱的[M+H]+模式在MRM下取得较好的响应,而且比较稳定,因此选择[M+H]+模式,这与宫旭等[21]报道的一致。根据马微[3]和李涛[4]等报道,酚酞采用正负离子模式都可检测,马微等[3]认为正离子响应高于负离子模式,但是本研究发现负离子的质谱响应信号高于正离子模式下的响应。在与其他药物同时检测时,采用正负离子同时扫描的模式,响应值明显低于正离子扫描模式。所以综合考虑,在流动相中加入10 mmol/L乙酸铵溶液,使得正离子条件下的酚酞也获得了较高的灵敏度,形成m/z 319的[M+H]+的准分子离子峰。对于结构相似的物质如洛伐他汀和辛伐他汀,裂解成相同的子离子,但通过离子对的组合,可以实现良好的分离。总之,通过优化各自的离子对,所有待测药物都选择了合适的母离子和子离子,部分药物的二级质谱图见图1。
2.2 色谱条件的选择
2.2.1 色谱柱
根据文献报道[20-22],结合选定的目标化合物的性质,分别考察了Agilent Eclipse Plus C18柱(2.1 mm×100 mm,1.8 μm)、Waters BEH Shield RP-18(100 mm×2.1 mm,1.7 μm)色谱柱、Kinetex 100 RP-18柱(100 mm×2.1 mm,2.6 μm)等。结果表明Agilent Eclipse Plus C18柱分离时整体出峰较好,但麻黄碱、咖啡因色谱峰出现拖尾;Waters BEH Shield RP-18柱适合分离碱性强的物质,因此麻黄碱、咖啡因峰形更尖锐对称,但是分离时酚酞和苯乙双胍峰不能分离;Kinetex 100 RP-18柱对8 个物质都能完全分离,各峰峰形也很尖锐对称,分析时间较短。综合考虑,最终选择Kinetex 100 RP-18柱(100 mm×2.1 mm,2.6 μm)为本实验的色谱柱。
2.2.2 流动相
对于反相色谱,通常采用甲醇-水、乙腈-水等二元溶剂体系作为流动相进行实验,根据许立等[9]报道,正离子扫描的模式通常采用甲酸增强仪器响应,但是也有文献采用加入不同比例的甲酸铵或乙酸铵取得很好的效果[21]。所以本研究分别比较了不同的溶剂配比对分离效果的影响,流动相中依次加入甲酸、乙酸、甲酸铵、乙酸铵等离子化试剂进行流动相的选择。结果发现:以不同体积比的乙腈-水为流动相时待测物的仪器响应信号高于同比例的甲醇-水为流动相。流动相中加入乙酸铵可改善待测物的分离与峰形,正离子模式中加入甲酸可显著提高待测物电离效果。这与许立等[9]的结果一致。所以最终选定乙腈和1 mmol/L乙酸铵(含1%乙酸)溶液作为流动相。在选定的流动相下各组分获得良好的分离,通过优化梯度洗脱程序,将不同极性的待测物的检测时间缩短为10 min。8 种待测物的总离子流图见图2。
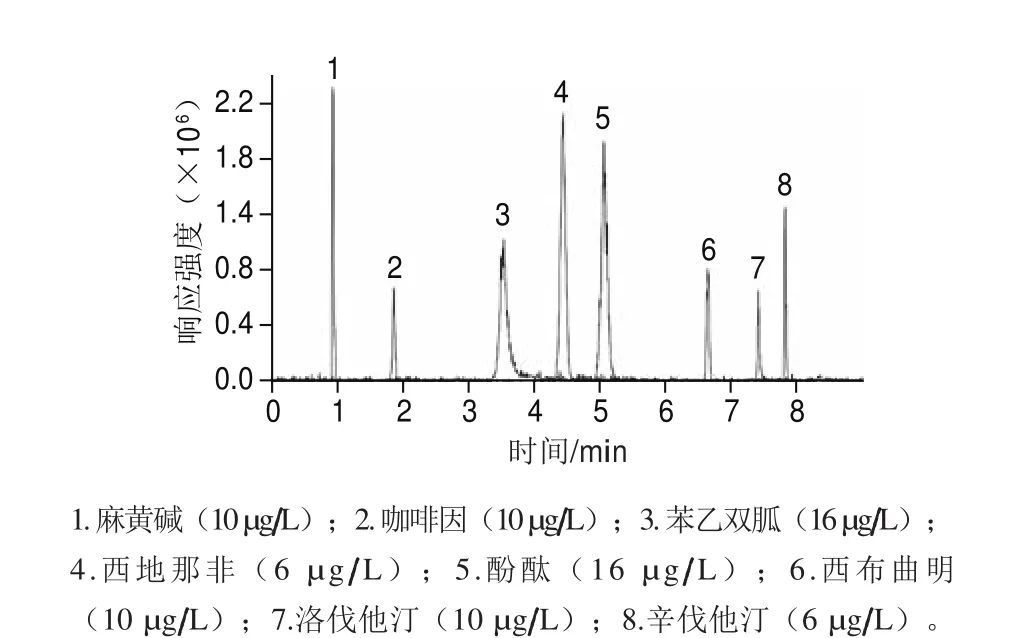
图2 8 种待测物的总离子流图(6~16 µg/L)Fig. 2 Total ion current chromatograms of the 8 compounds (6–16 µg/L)
2.2.3 柱温的选择
考察15、25、35、45 ℃ 4 种不同柱温对分离度的影响。通过比较发现在4 种温度条件下,分析总时间相差不大,但是在15 ℃时,洛伐他汀和辛伐他汀分离度下降,两峰会部分重叠。当柱温大于40 ℃时,苯乙双胍峰和酚酞峰因保留时间间隔缩短而未能分离,从而两种物质合为一个色谱强峰。柱温35 ℃时的实验结果最佳(图3)。因此本实验柱温最终定为35 ℃。
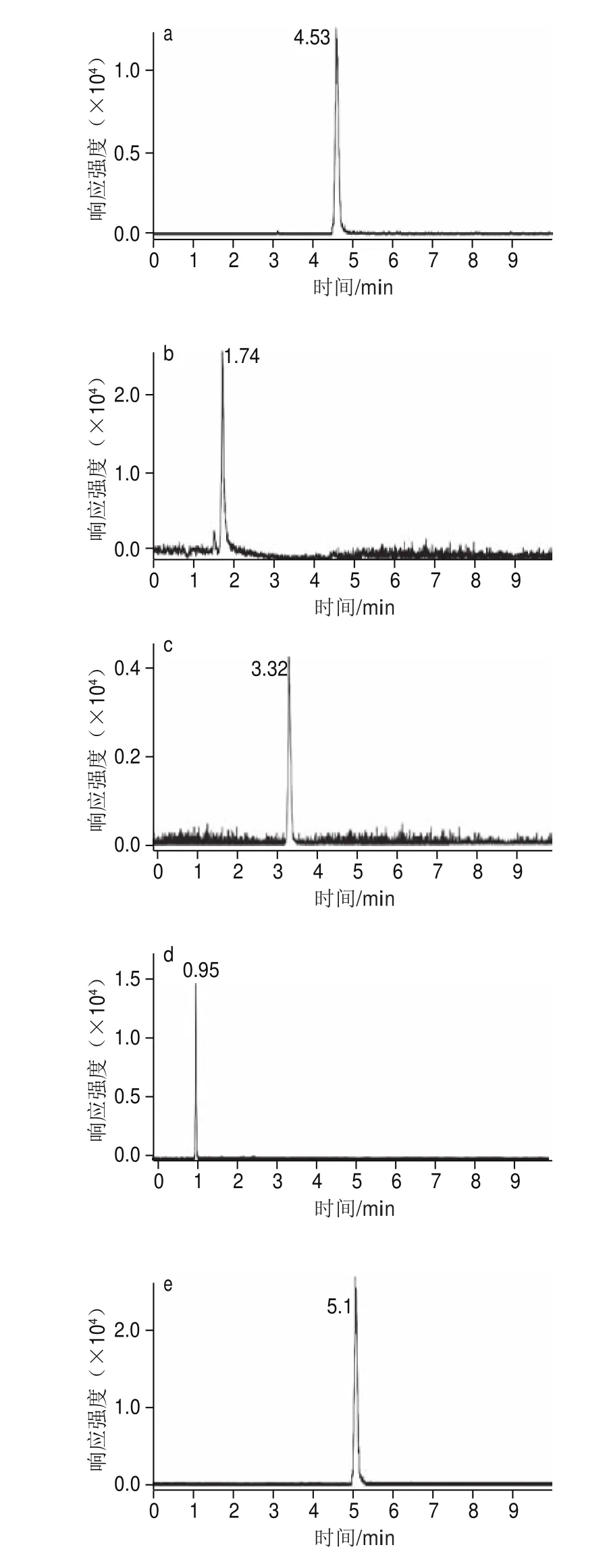

图3 标准溶液中8 种化合物的定量色谱图(0.6~1.6 µg/L)Fig. 3 Quantitative chromatograms of standard solutions of the 8 compounds (0.6–1.6 µg/L)
2.3 前处理方法的选择
直接提取法和SPE法分别参考文献[21]和文献[17-18]进行实验(如1.3.5节所述)。TLC法在文献[17-18]的基础上,对展开剂进行了优化,分别考察氯仿-甲醇-丙酮-冰醋酸(9∶2∶1∶0.5,V/V)和氯仿-甲醇-氨水(11∶1∶0.1,V/V)的展开效果,结果无明显差异。考虑到配比的方便,最终确定采用的是氯仿-甲醇-氨水(11∶1∶0.1,V/V)作为展开剂(具体步骤见1.3.5节)。
对3 种提取方法的提取效果进行详细比较,直接提取方法操作简单快速,但是稀释了药物的浓度,会影响测定的灵敏度,而且对于一些复杂的样品基质,只进行简单的提取,未经净化,上机样液杂质较多,容易导致色谱柱压力波动,影响保留时间定性,影响数据的准确性,而且极易污染离子源和检测器[27]。SPE法在提取后进行了净化,采用SPE小柱进一步对样品进行处理,消除了基质干扰,但是SPE成本高且操作较为复杂。此外文献认为SPE会降低方法的回收率[27]。所以本研究将色谱分析中的TLC法应用到样品前处理中,既避免了传统的液液萃取法容易造成干扰大、漏检、假阴性的结果,也摒弃了SPE成本高且较为复杂的方式。通过对3 种提取方法提取回收率的比较(表3),结果显示,采用TLC法前处理与SPE法取得同样的提取效果,明显优于传统的直接提取法,但TLC法比SPE法成本更低,操作更简单,更易于实现大规模的检测,最终本实验采用TLC进行前处理取得较好的效果。

表3 3 种不同的前处理方法比较Table 3 Comparison of three extraction techniques
2.4 方法学考察结果
2.4.1 基质效应考察结果
采用t检验考察基质效应[30]。经实验得出8 种药品在不同基质中的t值均大于2.4。所以,本研究采用基质匹配工作曲线进行定量。
2.4.2 检出限、定量限及线性范围
本研究在文献[20-22]的基础上,利用配制成的空白样品中加入混合标准溶液,按0.5、1.0、5.0、10、50、100 µg/L的水平添加,进样检测,绘制标准工作曲线。以峰面积Y对进样质量浓度X进行线性回归,以RSN为3计算检出限,以RSN为10计算定量限)。由表4可知,8 种药物在质量浓度0.5~100 μg/L范围内,线性关系良好,各组分的检出限在0.09~0.29 μg/kg之间,定量限在0.31~0.79 µg/kg之间。
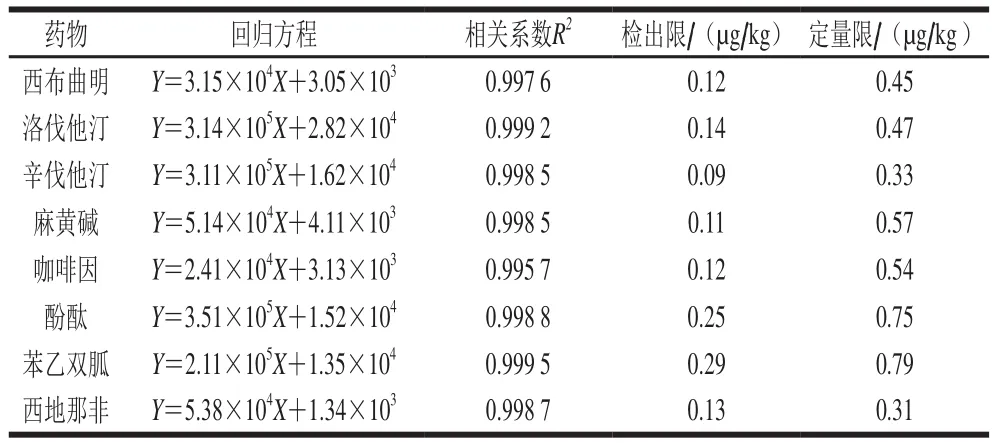
表4 8 种待测药物的线性回归方程、相关系数、检出限及定量限Table 4 Regression equations, correlation coefficients, limits of detection and limits of quantization for the 8 drugs
2.4.3 准确度和精密度结果
准确度用回收率表示,分别在3 个质量浓度水平上添加(1、2、4 倍定量限)。UPLC-MS/MS方法的专属性非常强,同类型样品基质的干扰可忽略,因此,本研究选取胶囊剂和口服液和其他固体制剂的代表粉末剂分别做回收率实验。如表5所示,对于胶囊剂和粉末剂,取其中阴性样品(12号),称取1 g样品,精密加入对照品储备溶液,最终使试样中西布曲明、洛伐他汀、麻黄碱、咖啡因含量0.5、1.0、2.0 μg/kg;辛伐他汀和西地那非含量为0.3、0.6、1.2 μg/kg,酚酞和苯乙双胍含量为0.8、1.6、3.2 μg/kg。对于口服液,取其中阴性样品(8号),量取1 mL样品,精密加入对照品储备溶液,最终使试样中西布曲明、洛伐他汀、麻黄碱、咖啡因含量为0.5、1.0、2.0 μg/kg;辛伐他汀和西地那非含量为0.3、0.6、1.2 μg/kg,酚酞和苯乙双胍含量为0.8、1.6、3.2 μg/kg。对于软胶囊剂,取其中阴性样品(43号),称取1 g样品,精密加入对照品储备溶液,最终使试样中西布曲明、洛伐他汀、麻黄碱、咖啡因含量为1.0、2.0、4.0 μg/kg;辛伐他汀和西地那非含量为0.5、1.0、2.0 μg/kg,酚酞和苯乙双胍含量为1.5、3.0、6.0 μg/kg。
按照前面优化的前处理方法和分析测定方法操作,每个含量样品重复5 次,通过连续5 d进行同样实验,以回收率表示方法的准确度,变异系数表示方法的精密度。不同样品中添加8 种药物的回收率及变异系数见表5,分析数据可知待测物在不同样品中的平均回收率介于67.3%~101.1%,变异系数≤16.5%,这些数据均符合分析方法标准对准确度和精密度的要求。
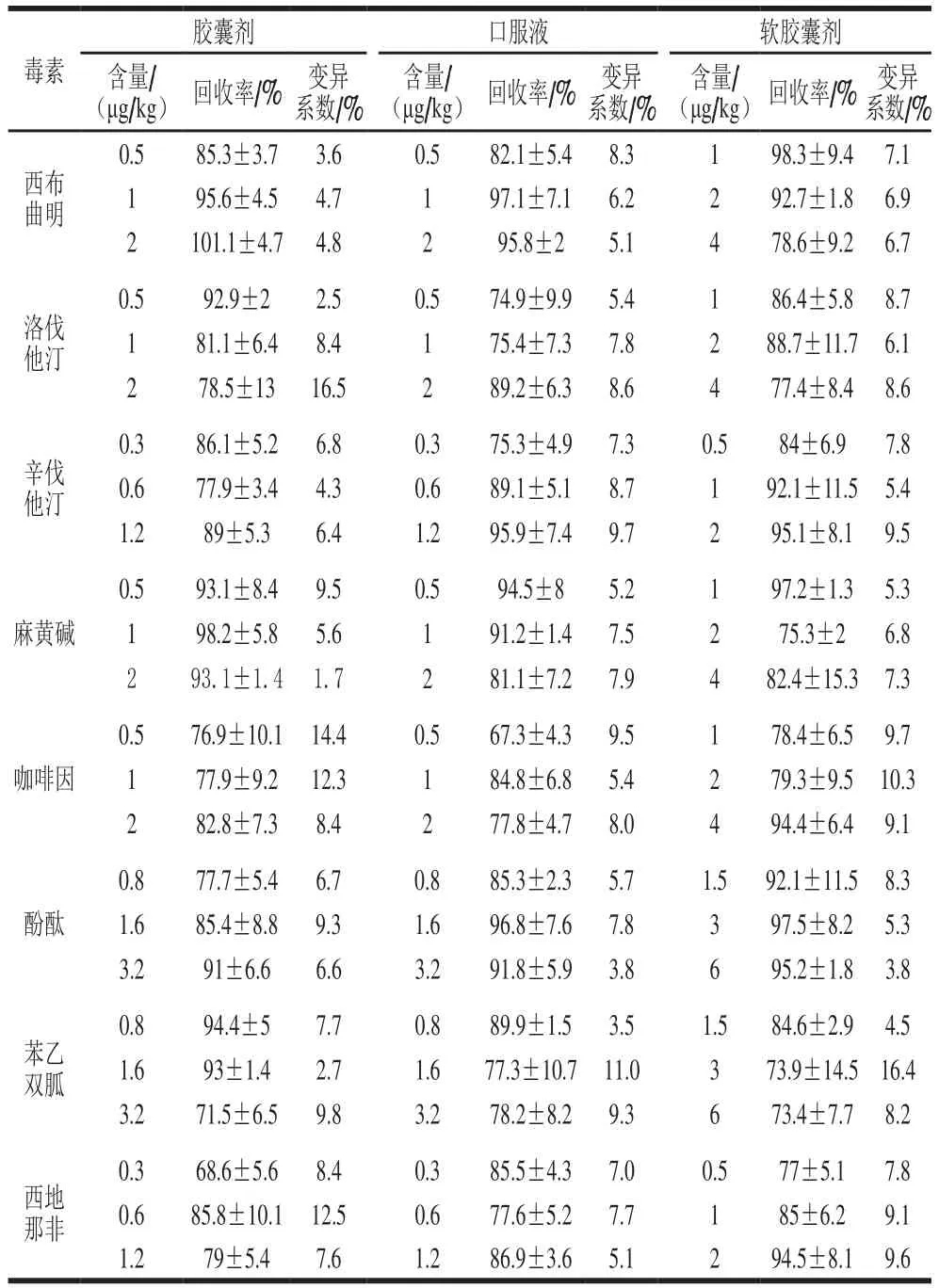
表5 不同样品中添加8 种药物回收率和变异系数Table 5 Recoveries and coefficients of variation for the 8 drugs from different spiked samples
2.5 样品测定结果
取网购保健食品样品,按1.3节制备方法制备,分别进样测定,根据定量监测离子对的离子流图中色谱峰面积,采用外标法计算样品中各化学成分的量。排除目标药物是由处方成分引入的,最后判定样品中是否含有非法添加的药物。
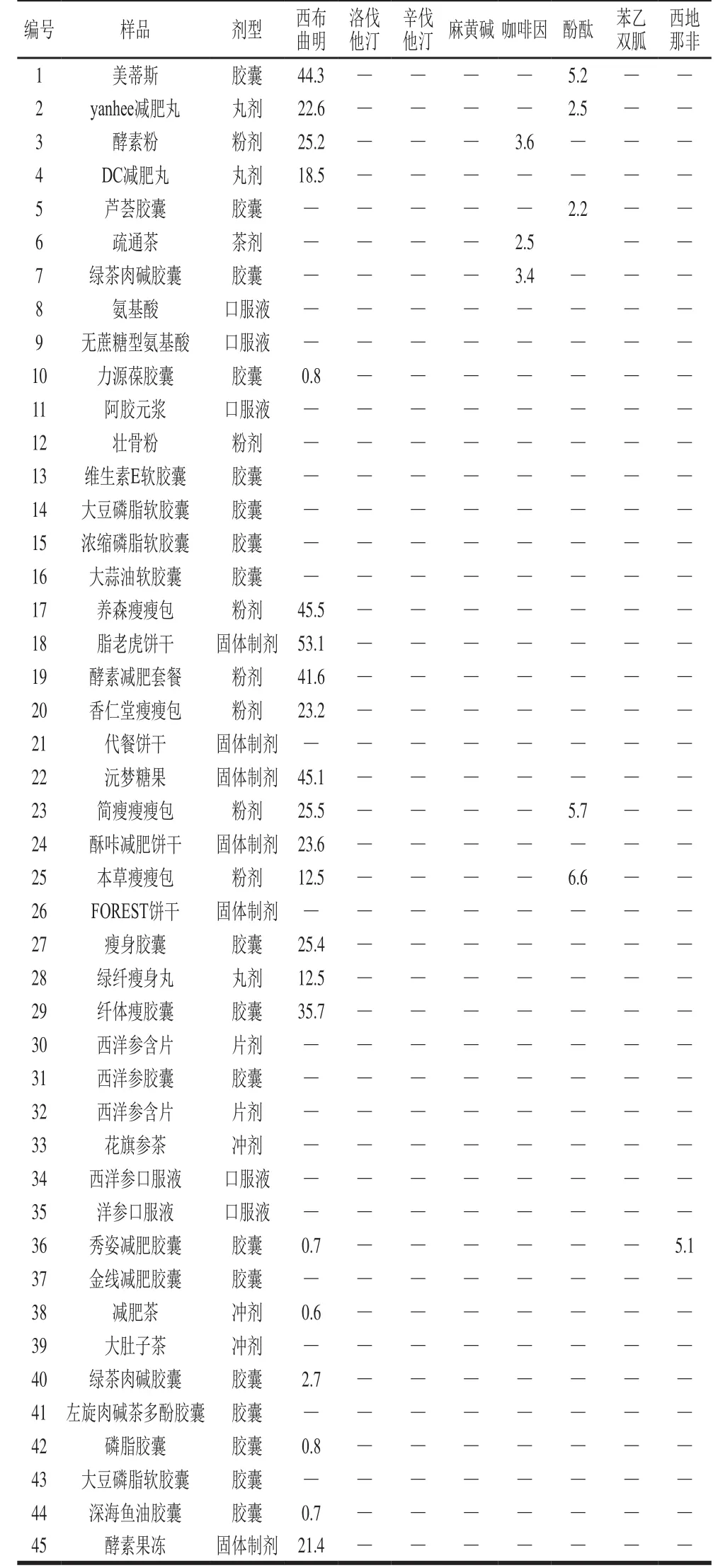
表6 45种网购样品中8 种待测药物的分析结果(n=6)Table 6 Results of detection of 8 drugs in 45 real samples (n= 6)
表6结果表明,45 批保健食品样品中25 批样品检出含有目标药物。通过与样品相应的成分进行比对,其中2 批样品中所检出的目标药物与处方固有成分一致,分别为疏通茶和绿茶肉碱胶囊检出咖啡因。因为茶叶中会有一定量的咖啡因,不属于非法添加;另外23 批样品检出的目标化合物均不属于处方所标示的成分,确定为非法添加阳性样品,采用2.5节线性方程计算其中非法添加药物的含量,结果表明:检出率较高的是西布曲明(21 批,含量为0.6~53.1 mg/g),酚酞(5 批,含量为2.2~6.6 mg/g),咖啡因和西地那非分别有1 批。
3 结 论
本研究建立了TLC-UPLC-MS/MS法检测网红保健品中非法添加的8 种化学药物的检测方法。根据方法学考察结果,本方法专属性强,灵敏度高,可作为非法掺入化学成分的有效检测方法。通过对网购的45 种网红保健品进行检测,为打击网络非法添加,规范保健品市场,提供了技术支持。
猜你喜欢
发明与创新·中学生(2017年12期)2017-12-11 20:27:29
发明与创新(2017年46期)2017-12-07 02:25:17
大自然探索(2017年10期)2017-10-28 06:47:59
大自然探索(2017年5期)2017-05-26 17:48:07
实用临床医学(2016年8期)2016-06-07 01:28:16
中国卫生标准管理(2015年17期)2016-01-20 09:26:45
发明与创新(2015年18期)2015-12-26 10:14:46
发明与创新·中学生(2015年5期)2015-06-08 00:30:50
中国当代医药(2015年17期)2015-03-01 02:03:39
中国卫生标准管理(2015年13期)2015-01-26 21:05:38
