
有机-无机杂化钙钛矿材料的本征稳定性*
2019-09-04 07:13张钰周欢萍
物理学报 2019年15期
张钰 周欢萍
(北京大学工学院,北京 100871)
1 引 言
人类社会自进入21世纪以来面临越来越严峻的化石能源枯竭、环境恶化和全球变暖等关系到人类存续的世界性难题,人类社会日益增长的能源需求与不可再生的传统化石资源之间的供需失衡将成为人类社会发展面临的主要矛盾.为了解决这一系列的问题,开发新型清洁可再生能源迫在眉睫.太阳能作为一类常见的可再生能源具有清洁无污染、储量丰富和就地利用等特点,有潜力成为下一代的支柱能源.通过太阳能电池将光能直接转化为电能是太阳能的主要利用方式之一,如何使太阳能发电变得更加低成本、高效能是世界范围内科学研究的热点领域.自1954年贝尔实验室首次成功研制单晶硅太阳能电池以来[1],硅基太阳能电池得到了巨大的发展和广泛的商业化应用.但是,硅基太阳能电池的制备工艺相对复杂,生产成本居高不下.为了进一步降低成本和扩大市场规模,科学家们一直在寻找性能更优异或成本更低廉的新材料太阳能电池,比如砷化镓(GaAs)、碲化镉(CdTe)、铜铟镓硒(CIGS)、铜锌锡硫(CZTS)等非硅无机半导体太阳能电池[2],以及染料敏化太阳能电池、有机太阳能电池等第三代太阳能电池[3].但是,新型的太阳能电池很难兼具高效、低成本等特点,目前为止并未撼动硅基太阳能电池在商用化市场上的统治地位,而近年来火热的钙钛矿太阳能电池为这个领域注入了新的活力和更多的可能性[4].
有赖于有机-无机杂化钙钛矿材料优异的光电性质,自2009年首次将其作为吸光层引入太阳能电池以来[5],钙钛矿太阳能电池的光电转换效率在短短十年间由最初的3.8%飞速提高到了24.2%[6],仅从效率的角度来看已经接近单晶硅太阳能电池.基于钙钛矿材料本征的优异光电特性,研究人员通过组分调节、体相及界面缺陷钝化、器件结构优化、光管理等一系列手段实现了效率的快速提升[7−11].
尽管钙钛矿太阳能电池在效率上发展突飞猛进,但是这种材料的不稳定性严重阻碍了它走向商业化的脚步[12,13].最初的钙钛矿太阳能电池继承了染料敏化太阳能电池的器件结构即液态电解质型器件,钙钛矿材料较强的反应活性导致它在液态电解质中稳定性很差,致使器件性能快速蜕化.Kim等[14]将固态空穴传输层引入钙钛矿太阳能电池中从而得到了全固态器件,这样一来钙钛矿太阳能电池的光电转换效率和稳定性均得到了显著的提升,也由此掀起了钙钛矿电池领域的研究热潮.虽然固态器件的结构大体被沿用下来,但是器件结构带来的不稳定性因素只是一个方面,有机-无机杂化钙钛矿材料的本征不稳定性才是问题的核心.钙钛矿材料的本征不稳定性主要表现在当它暴露在一定程度的湿度、光照或加热的条件下会出现自发的蜕化从而导致器件性能的不可逆下降.钙钛矿材料本征不稳定性有诸多原因,一方面钙钛矿具有离子键合特性并且组成离子均为离子势较小的“软”离子,且含有较易分解的有机铵离子,这使得钙钛矿体系形成能较小、缺陷密度较高、各组分反应活性大,从而存在化学稳定性较差的问题.此外,温度或者组分的差异会导致钙钛矿材料发生成分偏析或者相分离,这是钙钛矿及钙钛矿太阳能电池不稳定性的另一重要方面.因此,本文从化学不稳定性和相不稳定性这两个基础层面剖析了有机-无机杂化钙钛矿材料的本征不稳定性的问题,并梳理了近年来针对改善各项不稳定性所做的努力,从组分设计及制备工艺等角度提出了提高钙钛矿太阳能电池器件稳定性的可能的途径.
2 有机-无机杂化钙钛矿材料的化学不稳定性
有机-无机杂化钙钛矿材料的化学不稳定性是指这类钙钛矿材料在环境及化学因素的作用下,如水、氧、光照以及加热等条件下无法保持原有结构及物理化学性质的行为及现象.研究人员尝试了多种手段来增强钙钛矿材料的化学稳定性,比如组分工程、添加剂工程、结构设计、封装工艺等.本节讨论化学不稳定性的内因和表现,并结合提升钙钛矿材料化学稳定性的最新研究进展,进一步分析影响化学稳定性的内在机理和可能的解决方案.
2.1 热致化学不稳定性
钙钛矿太阳能电池作为一款面向商业化的新一代低成本高效率光伏器件,必须要能够承受由光照引起的一定的高温工作环境的考验.按照IEC(International Electrotechnical Commission)标准,钙钛矿太阳能电池模组在进入市场之前必须要通过 85 ℃ 条件下的高温老化测试.因此,克服钙钛矿材料的热致化学不稳定性就显得至关重要.有关研究表明,CH3NH3PbI3(MAPbI3)体系的形成能仅有 0.11 eV 到 0.14 eV,这与 85 ℃ 对应的能量 0.093 eV(3KbT)相当,因而 MAPbI3体系存在显著的热不稳定性[15].实验证据也表明,MAPbI3在100 ℃ 左右持续退火便可以观察到按下述方程式显著的分解[15,16],

MAPbI3体系较差的热稳定性主要源自于甲铵离子.MA+中的C—N键和N—H键均为极性较强的共价单键,这种类型的化学键键能小,容易在较高的温度下断键分解,而A位离子的分解会带来钙钛矿整体结构的不可逆崩塌[17].有鉴于此,研究人员开发了更加耐热的A位阳离子及其相应的钙钛矿太阳能电池.与MA+相比,含有大 π 键并且正电荷离域化的甲脒离子FA+以及无机离子Cs+,Rb+显然具有更优的热稳定性(图1)[18].

图1 MA+,FA+,Cs+,Rb+结构示意图[18]Fig.1.Schematic diagram of MA+,FA+,Cs+,Rb+[18].
Eperon 等[19]发现,在 180 ℃ 的退火温度下FAPbI3在60 min内都能够保持很好的稳定性,而MAPbI3在同样条件下则完全分解为碘化铅.Lee等[20]也证明了在175 ℃以下未观察到FAPbI3的任何分解行为.而Cs+由于其离子半径过小,具有光电活性的立方相CsPbI3钙钛矿结构在室温下难以稳定存在,围绕该体系所做的研究主要是针对相稳定性,故一般不探讨CsPbI3体系的热稳定性问题[21−24].此外,FAPbI3在室温下还存在容易从黑色a相转变为黄色d相的问题[25].研究人员发现通过混合FA+和Cs+得到的二元钙钛矿体系,既能解决相不稳定的问题,又保证了很好的热稳定性[26].在此基础上,Turren-Cruz 等[18]向二元体系中进一步引入Rb+得到的RbCsFA三元体系不仅光电转换效率达到了20%以上,而且有着很好的热稳定性和工作状态下的长期稳定性.
2.2 光致化学不稳定性
太阳能电池在工作状态下需要接受来自太阳光谱各波段的照射,而光被吸收并转换为电能的同时,还可能会对材料的结构和化学性质造成损伤,尤其是波长较短、能量较高的紫外光更是钙钛矿光不稳定性的重要来源.研究表明,MAPbI3在光照和干燥氧气的存在下会按下式所示的机理分解为甲胺、PbI2和I2[27]:

简单来说,在光照下钙钛矿产生的光生电子与环境中的氧气发生电子转移从而生成了超氧负离子,而超氧负离子具有很高的氧化性和不稳定性,从而进一步与钙钛矿中的有机阳离子反应使钙钛矿彻底蜕化为甲胺、PbI2和I2.甲铵离子的质子酸属性在一定程度上促进了反应的进行,因此研究人员建议将A位阳离子更换为非质子型阳离子.
与MA+相比,尽管FA+也是质子型阳离子,但是由于FA+中存在的电子共轭体系带来了额外的稳定化能,使得基于FA的钙钛矿体系光不稳定性得到了很大改善.前述提到的无MA的RbCsFA三元体系在光照下可以在最大功率点稳定输出1000 h而不会显著衰减(图2).

图2 RbCsFA三元钙钛矿太阳能电池最大功率点输出[18]Fig.2.Maximum power point tracking of RbCsFA hybrid perovskite solar cells[18].
除了有机阳离子所带来的光不稳定性因素之外,紫外光还会直接破坏Pb—I离子键,导致零价金属 Pb0和卤素单质 I0的形成[28−30].Pb0的形成会给体系带来深能级缺陷,从而极大地损害器件的性能和长期稳定性[31,32],而I0的形成也会成为载流子的复合中心,并可能引发链式反应催化钙钛矿的分解[28].已经报道了几种尝试消除Pb0和I0缺陷的方法,主要是通过优化薄膜工艺和添加剂工程的手段,比如优化空气中的后退火工艺来降低Pb0的含量[33],利用还原性次磷酸消除I0缺陷[34],也有利用碱性环境下的歧化反应来消除I0缺陷[35]等.然而这些方法均不能在器件长期的工作状态下持续抑制铅碘零价缺陷的产生,所以对钙钛矿的长期稳定性提升是比较有限的.最近,Wang等[36]针对这个问题提出了一种全新的解决思路,即通过变价金属Eu3+/Eu2+的掺杂,实现了对两种零价缺陷的催化自修复.其消除零价缺陷的机理如图3所示.

图3 Eu3+/Eu2+催化消除铅、碘零价缺陷机理[36]Fig.3.Proposed mechanism diagram of cyclically elimination of Pb0 and I0 defects and regeneration of Eu3+-Eu2+ metal ion pair[36].
Eu3+/Eu2+离子对在整个反应过程中扮演了可循环的氧化还原梭的角色,因而在钙钛矿太阳能电池的全寿命周期都能够起作用,是帮助实现钙钛矿太阳能电池长期稳定性的一种新颖且有效的解决方案(图3).
2.3 水致化学不稳定性
钙钛矿太阳能电池在潮湿环境下的蜕化是一个极具挑战性的问题,而组成钙钛矿的铵盐的吸湿性是钙钛矿材料在潮湿环境下不稳定的主要原因.钙钛矿材料强烈的吸水性使得这种材料可以吸附环境中的水分子从而形成(CH3NH3)4PbI6·2H2O等形式的水合物[37],该类水合物的形成会对器件的性能造成损害.而在水氧同时存在的条件下,钙钛矿还会按照下式产生不可逆的分解[38]:

水首先溶解MAPbI3形成CH3NH3I的溶液和PbI2,而CH3NH3I溶液将继续分解成CH3NH2溶液和HI溶液.在氧气存在下,HI可以很容易被不可逆氧化成I2,H2O,并且光照会进一步加速这种氧化过程.Walsh研究组[39]还提出了一种水分子催化分解途径,他们认为单个水分子足以导致钙钛矿材料的降解.随着钙钛矿的分解,过量的水会溶解副产物HI和CH3NH2,直到HI在H2O中达到饱和或CH3NH2的蒸气压达到平衡,这使得在封闭系统中痕量的H2O就可以导致钙钛矿的严重分解.在足量水存在的情况下,钙钛矿材料将完全分解形成PbI2.从机理来看,有机阳离子的质子酸属性起着决定性的作用,因此如果改变或者削弱有机阳离子的质子酸属性,理论上钙钛矿材料的水稳定性会得到极大的提升.
通过组分工程的努力,人们发现向钙钛矿体系中引入具有疏水尾端的有机胺阳离子可以获得湿稳定性远胜于三维钙钛矿的准二维钙钛矿材料[40−43].Smith 等[44]采用组分为 PEA2MA2Pb3I10(PEA=C6H5(CH2)2NH3+)的准二维钙钛矿材料作为吸光层(图4),在环境条件下通过旋涂获得了高质量的钙钛矿薄膜,即便不经过进一步的退火,也比常规的三维钙钛矿薄膜更耐潮湿.这主要得益于准二维钙钛矿结构更高的形成能和疏水有机插层对水分子的排斥作用.粉末X射线衍射(XRD)的结果显示,在潮湿的环境下储存46 d后,相比于已经完全分解为碘化铅的三维钙钛矿材料,二维钙钛矿材料的特征衍射峰的相对强度没有太大的变化.
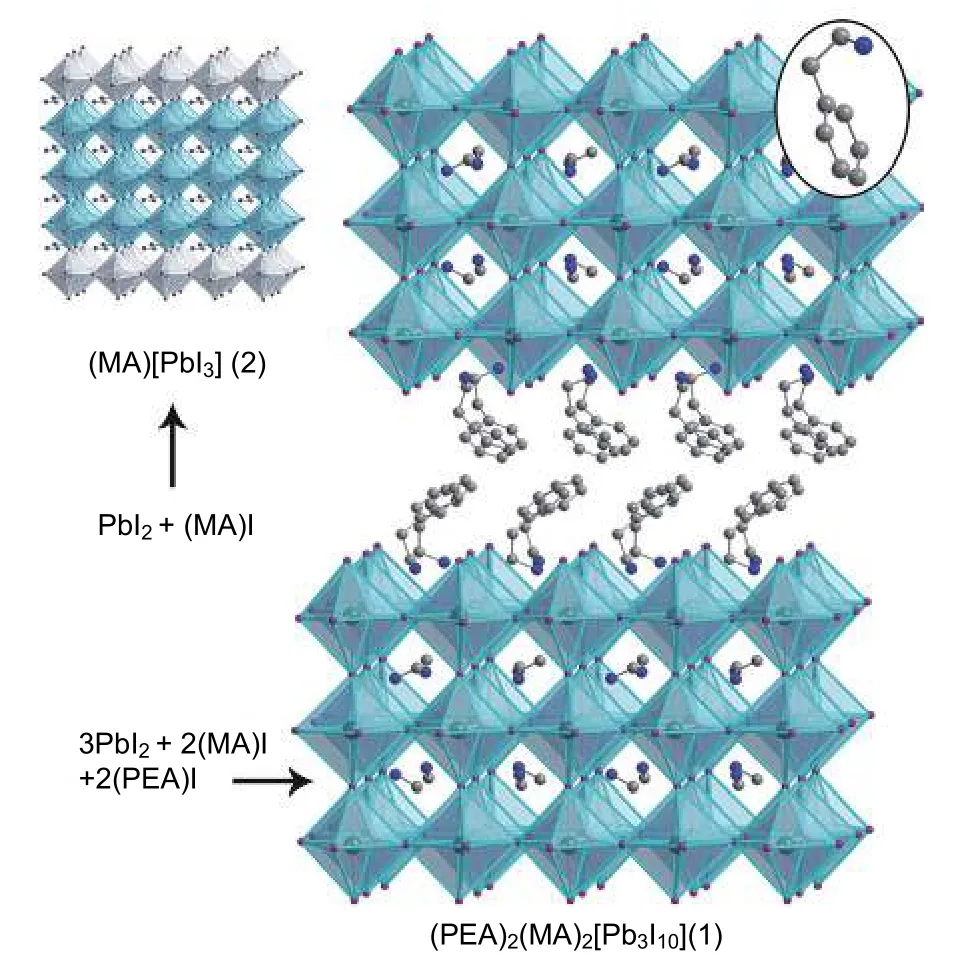
图4 PEA2MA2Pb3I10 晶体结构示意图[44]Fig.4.Crystal structure of PEA2MA2Pb3I10[44].
然而准二维钙钛矿中存在的量子阱结构会使得激子结合能增加并增强晶格对载流子的散射,所以基于准二维钙钛矿的太阳能电池光电转换效率无法与一般钙钛矿太阳能电池匹敌.有鉴于此,研究人员开发了一系列方法来将稳定性好的二维钙钛矿材料与效率高的三维钙钛矿相结合[45].比如Li等[46]利用竖直沉积在三维钙钛矿薄膜上的纯二维钙钛矿来达到修饰和钝化的目的,最终在保持了高效率(>21%)的同时也获得了非常好的稳定性,相应器件在空气中3000 h后仍保持了初始效率90%的效率输出.
从化学的角度出发,引入功能性分子或者基团,在不改变钙钛矿组分的前提下同样可以提升钙钛矿材料的湿稳定性.Zhao等[47]通过向钙钛矿中引入聚乙二醇(PEG)形成的高分子网络结构,使钙钛矿获得了很好的湿稳定性,相应的器件在70%的湿度环境中存放300 h仍保持了约70%的效率输出.这主要是由于PEG的强吸湿性束缚住水分子从而抑制了水对钙钛矿的分解作用.与此同时,PEG与甲铵离子的强束缚作用使得在湿度环境下已经解离的甲铵离子无法逸出,离开湿度环境之后甲铵离子重新与碘化铅反应生成钙钛矿,从而实现了“自愈”性.Yang等[48]通过在钙钛矿表面引入一系列的疏水叔铵或季铵离子有效地提高了钙钛矿的湿稳定性,表面修饰四乙基铵离子的钙钛矿太阳能电池在90%的湿度下储存24 h后效率仍能保持初始效率的90%.
2.4 缺陷与离子迁移
针对金属晶体和原子晶体提出的经典缺陷理论往往按照缺陷在空间中的延伸维度不同分为点缺陷(空位缺陷、填隙缺陷、杂质原子等)、线缺陷(刃型位错、螺型位错等)、面缺陷(堆垛层错、晶界、相界、孪晶界等)和体缺陷(气孔、第二相夹杂物等)四种类型.而有机无机杂化钙钛矿材料是一类通过库仑相互作用形成的离子晶体,因此在讨论钙钛矿材料中的缺陷时必须要结合离子晶体的特殊性.接下来我们结合这种特殊性详细讨论钙钛矿材料中的点、线和面缺陷.
有机-无机杂化钙钛矿具有迥异于传统无机半导体材料的离子性成键方式,并且各组分所带的电荷量少、离子半径大,故而相应的离子键键能低,晶体内部易产生各类点缺陷,甚至各组分含量易偏离化学计量比而形成非化学计量比钙钛矿.文献[49−53]分别报道了钙钛矿晶体内存在的点缺陷并结合计算模拟给出了各类点缺陷在能带结构中所占据的相应缺陷能级,如图5所示.

图5 MAPbI3 中各类型点缺陷能级位置[49]Fig.5.Calculated transition energy levels of point defects in MAPbI3[49].
可以看出这其中大部分缺陷类型属于浅能级缺陷,还有相当一部分的缺陷能级并不位于带隙范围内,只有铅、碘反占位缺陷(PbI,IPb)、铅填隙缺陷(Pbi)以及碘占据MA位缺陷(IMA)四种深能级缺陷,而有关计算表明深能级缺陷的形成能很高,在材料中的含量非常少.因此,即便钙钛矿材料的总体缺陷浓度很高,但由于绝大部分缺陷所处能级并不位于带隙深能级,使得钙钛矿材料拥有着超高的缺陷态容忍度.针对存在的各类点缺陷,研究人员也开发了一系列的钝化手段和薄膜制备工艺以减小体相中由于点缺陷造成的非辐射复合[54],比如Snaith研究组[9]发现在钙钛矿前驱液中加入少量的MACl可以显著降低缺陷态密度并提高MAPbI3的晶体质量和载流子扩散长度;Park和Seok[53]发现基于PbI2-DMSO分子内交换的两步法制备钙钛矿时存在一种碘缺乏的中间产物[(Pb3I8)n]2n–,从而可能导致钙钛矿中深能级缺陷的形成.他们发现通过向两步法IPA溶液中引入额外的I3–,能够有效抑制相关缺陷的形成,并获得了当时最高的22.1%的认证效率[55];Chen等[56]通过在前驱液中引入碱性物质,促使单质碘杂质在不同的碱性环境下发生歧化反应,有效地消除了前驱液中的单质碘杂质并大幅降低了钙钛矿薄膜中深能级缺陷的密度;Saliba等[57]将Rb+掺杂到钙钛矿中发现对器件的高效率和稳定性都有很大的提升,并且其器件的开压高达 1.24 V,这说明 Rb+的掺杂有效降低了缺陷态密度从而抑制了非辐射复合的发生.Yang等[58]发现带有双羰基的共轭路易斯碱咖啡因与Pb2+之间的强烈相互作用增加了钙钛矿薄膜的生长活化能从而促进了高质量薄膜的生长,抑制了离子迁移并大大提高了钙钛矿的热稳定性;Lee等[59]选择了一种弱极性、无毒、化学惰性的全氟萘烷作为新的退火介质,通过延缓溶剂挥发有效延长了晶体的生长时间,使得晶粒得到显著增大并降低了缺陷态密度.此外,如2.2节所提到的在光照或者加热状态下产生的零价铅、碘缺陷也会对器件性能造成极大的损害,针对这种工作状态下不断产生的缺陷,Wang等[36]提出的利用Eu3+/Eu2+氧化还原梭催化零价缺陷复原是目前最行之有效的方法之一.
在钙钛矿中我们很少讨论线缺陷,这主要是由离子晶体和金属以及原子晶体成键的差异性导致的.线缺陷一般由滑移过程产生,这一过程对于无方向性和饱和性的金属键来说是易发生的,而对于有方向性和饱和性的离子键来说是不易发生的.因为滑移可能会使同性电荷相互靠近,从而破坏库仑力的平衡最终导致晶体碎裂,同时这也是离子晶体韧性较差的主要原因.尽管如此,在新切割的MAPbBr3的晶体表面通过扫描隧道显微镜观察还是能够发现线缺陷的存在[60],但目前我们对这一类型的缺陷与器件性能的关系知之甚少.
我们所讨论的钙钛矿中的面缺陷主要包括孪晶界、晶界和传输界面.Rothmann等[61]通过透射电子显微镜直接观察到了畴区宽度在100—300 nm的孪晶的存在,Liu等[62]证明了孪晶畴具有铁弹性以及潜在的化学偏析和弹性变化效应,并且这些现象的存在会影响器件的光电响应,但目前对于孪晶与器件性能的相互作用仍然没有确凿的认识.钙钛矿的晶界和与传输层之间的界面由于晶格断裂存在众多的悬挂键和复合中心,也是钙钛矿中离子迁移的主要通道[63],针对这两处界面开展钝化是提高钙钛矿太阳能电池性能的有效途径.比如Chen等[64]通过控制退火时间来调节钙钛矿表界面处的碘化铅的产生,发现一定量的碘化铅可以有效钝化钙钛矿表界面的缺陷从而提高器件的性能;Shao等[65]证明了表界面的缺陷态是钙钛矿太阳能电池存在迟滞的重要原因,他们发现通过在表界面引入富勒烯衍生物PCBM能够使表面缺陷态密度降低两个数量级,显著提高了器件性能并消除了迟滞;后来他们通过实验发现具有两性离子结构的季铵盐可以同时钝化钙钛矿表面存在的带正电性的路易斯酸性缺陷以及带负电性的路易斯碱性缺陷[66];他们还进一步对可能具有钝化效果的基团进行了详细的探究,发现氨基和羧基可以通过静电相互作用钝化钙钛矿表界面的离子型缺陷,而芳香基团可以抑制与零价碘相关的缺陷,据此引入经过设计的钝化分子 D-4-tert-butylphenylalanine(D4TBP)可以制备得到效率达21.3%的反式电池,开压高达 1.23 V[67].Tan等[68]针对传输界面的钝化开展了专门的研究,他们使用氯封端的TiO2胶体纳米颗粒作为传输层的同时取得了很好的界面钝化效果,在1.1 cm2的器件上获得了19.5%的器件性能.Abdi-Jalebi等[69]发现向钙钛矿的表界面引入一层卤化钾钝化层可以大幅缓解钙钛矿薄膜界面的非辐射复合和光诱导离子迁移;Wang等[70]发现苯乙胺分子中的共轭结构有利于电荷传输并且氨基与钙钛矿有较强的相互作用,再加上苯乙胺本身具有强疏水性,因此向钙钛矿表界面引入苯乙胺分子不仅达到了钝化缺陷的效果,还大大增强了钙钛矿的耐湿性.而Jiang等[71]通过旋涂的方式在钙钛矿表面引入了一层碘化苯乙铵,发现碘化苯乙铵对钙钛矿表界面也具有显著的钝化效果,利用这种方式获得的器件具有明显改善的开压损失,并获得了当时最高的23.32%的认证效率.
缺陷是离子迁移的重要通道,而钙钛矿中存在的大量缺陷使得离子迁移很容易发生[72].有计算表明,MA+,Pb2+和 I–的迁移活化能分别为 0.84,2.31和0.58 eV,说明碘离子是离子迁移的主体[73].离子迁移带来了许多危害,一方面它是造成钙钛矿太阳能电池存在“迟滞”现象的重要原因[74,75],另一方面,离子迁移的积累会造成钙钛矿晶体结构的崩塌从而极大地损害器件的长期稳定性.已经证明,在加热、外加偏压或光照等条件下离子迁移会变得十分严重[76].碘离子甚至能从多晶钙钛矿薄膜中迁移出来进而在相邻的界面产生缺陷甚至影响金属电极,这些新产生的缺陷作为非辐射复合中心对器件整体性能有非常不利的影响[77].为了改善器件的性能和长期稳定性,研究人员采取了大量的策略试图抑制离子迁移.近来的研究发现掺入钾离子(K+)可以有效地抑制缺陷的形成和离子迁移[78].Son等[78]的研究表明在钙钛矿前驱液中只需加入10 μmol的KI就可以达到降低缺陷密度并抑制离子迁移、降低迟滞的效果,并且这种添加对于不同组分的钙钛矿都具有相似的效果.他们认为其原因是钾离子占据了间隙位并阻止了弗伦克尔缺陷的形成.Abdi-Jalebi等[69]发现添加的钾离子实际上主要位于晶界处,可以起到钝化卤素离子空位和阻止离子迁移的作用.除此之外,准二维钙钛矿体系也有很好地抑制离子迁移的表现,这主要是因为长链有机铵离子组成的有机空间层有效地抑制了离子迁移[79].
3 相不稳定性
温度或者组分差异可能导致钙钛矿材料存在潜在的相不稳定性,这是钙钛矿材料本征不稳定性的另一重要原因.从晶体学的角度考虑一个组成为ABX3的立方钙钛矿结构,可以发现A位通常是一个尺寸较大的并且具有十二配位结构的阳离子,而B位通常是一个较小一些的具有六配位结构的金属离子,同时B与X形成的[BX6]配位多面体通过共点连接的方式形成了整个钙钛矿的三维骨架,而A位离子恰好填充在三维骨架的十二面体空隙中,起到了支撑骨架的作用.只有当每种离子的电荷和半径都满足结构的要求,才能形成钙钛矿结构的晶体.为了量化对钙钛矿结构的描述,Goldschmidt等[80]早在1926年就提出了尺寸容忍因子t的概念,其定义为:

其中rA,rB和rX分别代表A,B,X三种离子的离子半径.一个稳定的钙钛矿结构,其各组分离子半径的大小受尺寸容忍因子t的限制.最完美钙钛矿结构的t=1,而实际上当t介于 0.89—1 之间时都有潜力形成稳定或者亚稳的钙钛矿结构.研究人员通过计算模拟得出了APbI3中较为常见的三种阳离子的t值,虽然不同的研究组得出的结果略有不同,但总体趋势是类似的,综合来看t(Cs+)在0.8左右,t(MA+)介于0.91到0.99之间而t(FA+)稍大于1[81,82].由此可以看出尺寸容忍因子偏差较大的纯相CsPbI3和FAPbI3在室温下都无法形成稳定的立方钙钛矿结构,CsPbI3的立方相转变温度在590 K以上,而FAPbI3的薄膜在室温下容易发生相转变,生成六方相的黄色物质[24,25].MAPbI3的尺寸容忍因子虽然较为合适,但在常温条件下表现为四方相结构,在327.4 K的温度下才发生从四方相到立方相的转变[83].有鉴于此,采取混合阳离子策略以获取更优的尺寸容忍因子是极有必要的.
事实上,研究人员开发了多种性能优异、稳定性好的多元阳离子体系,比如FACs,FAMACs,FACsRb等[18,26,84].这些多元体系的特点就是拥有合适的尺寸容忍因子从而相稳定性较好,并且表现出的光电性能和长期稳定性也大大优于纯相的钙钛矿体系.而另一方面,多元钙钛矿材料体系由于其组分多样,不同组分具有不同的溶解性以及动力学/热力学特性,从而也可能面临相分离和成分偏析的问题.如对于FAMAPbIBr体系的钙钛矿材料来说,研究人员就观察到卤素离子在整个薄膜上存在成分偏析,分布并不均匀,这对于器件性能和长期稳定性是不利的.针对这一问题,最近Correa-Baena等[85]利用同步加速纳米级X射线荧光光谱等技术发现碱金属离子Cs,Rb的添加能够有效促进多元钙钛矿体系中的卤素特别是Br离子的均匀分布(图6),这启发我们可以通过组分工程和添加剂工程的手段有效抑制钙钛矿的相不稳定性.

图6 X射线荧光光谱显示出不同碱金属卤化物添加剂情况下的Br离子分布[85]Fig.6.X-ray fluorescence mapping indicates heterogeneous distribution of Br as a function of alkali metal incorporation of the perovskite films[85].
此外,全无机钙钛矿尤其是CsPbI3由于不含有有机离子,具有更优的化学稳定性,吸引了许多研究人员的兴趣.但由于Cs+过小,CsPbI3存在相不稳定性的问题,因此如何获得室温稳定的具有光学活性的立方或四方相结构成为该领域关注的热点.目前获得稳定CsPbI3室温光活性相结构的方法主要是通过组分工程、添加剂工程和成膜工艺这三种手段来实现的.组分工程主要是通过将钙钛矿中部分碘离子替换为溴离子从而使相结构稳定在光活性相态.比如Liu等[86]将1/3的碘替换为溴,获得了相结构稳定的CsPbI2Br钙钛矿材料,并且获得了超过13%的光电转换效率.添加剂工程主要是通过添加能够抑制CsPbI3相变的添加物来达到稳定相态的目的.比如有报道在钙钛矿前驱液中添加适量HI能够有效提高全无机钙钛矿的室温相稳定性[22,24],但Ke等[87]指出这主要是由于前驱液的溶剂二甲基甲酰胺(DMF)与部分HI反应生成的二甲胺离子(DMA+)参与形成了钙钛矿(图7).此外,Wang等[23]通过调节成膜工艺也成功获得了在氮气氛下室温稳定的全无机钙钛矿太阳能电池,但在潮湿的空气中会很快发生相转变.全无机钙钛矿要达到室温下的长期相稳定性还需要较大的努力,后续有希望通过组分工程和添加剂工程寻找到能够有效抑制相转变的方法.
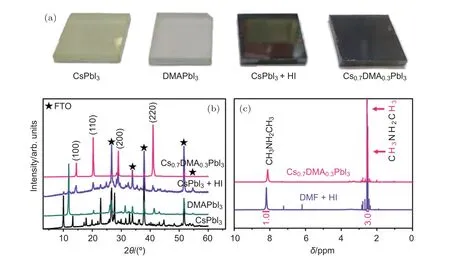
图7 (a)DMA 掺杂薄膜与 HI酸添加薄膜对比;(b)DMA 掺杂薄膜与 HI酸添加薄膜 XRD 对比;(c)DMA 掺杂薄膜与 DMF 和HI反应所得产物DMAI的核磁共振对比[87]Fig.7.Film properties and component studies:(a)Photographs,(b)XRD spectra,(c)nuclear magnetic resonance spectra of the Cs0.7DMA0.3PbI3 films and DMAI polycrystalline powder synthesized from DMF and HI[87].
4 结 论
有机-无机杂化钙钛矿材料作为光伏领域的后起之秀,引发了空前的研究热潮并受到了社会各界广泛的关注.钙钛矿太阳能电池最大的优势是它的光电转换效率高而成本相对低廉,但是钙钛矿材料的本征不稳定性却大大阻碍了它的商业化进程.为了推进这一问题的解决,我们从化学不稳定性和相不稳定性两个角度出发,对钙钛矿材料的本征不稳定性进行了较为详细的探讨.有鉴于MA+在化学稳定性的各个方面较差的表现,我们认为具备长期稳定性的钙钛矿太阳能电池应该极力减少MA+的使用.基于掺杂少量无机碱金属离子如Cs,Rb的FA主相的钙钛矿体系是目前理想的选择,但要进一步提高钙钛矿材料的稳定性势必要继续推进组分工程、添加剂工程、制备工艺等诸方面的研究.寻找化学性质更稳定、尺寸因子更合适的钙钛矿体系、开发类钙钛矿的稳定新材料体系以及通过化学手段提高钙钛矿稳定性等措施并举,将会有效推进钙钛矿太阳能电池的商业化进程.
猜你喜欢
无机化学学报(2022年8期)2022-08-09
无机材料学报(2022年1期)2022-04-12
燃烧科学与技术(2021年5期)2021-10-28
考试与评价·高一版(2020年2期)2020-10-29
现代养生·下半月(2017年9期)2018-01-04
中国高新技术企业(2017年8期)2017-06-05
科技视界(2016年15期)2016-06-30
北京信息科技大学学报(自然科学版)(2016年6期)2016-02-27
能源研究与信息(2014年3期)2014-10-30
电子世界(2004年5期)2004-07-26
