
基于全基因组RNA测序数据和基因集富集分析方法对直肠癌发病机制的初步探讨*
2019-09-03 09:41:10廖存廖锡文韦瑞丽黄伟龚艺贞马辉
结直肠肛门外科 2019年4期
廖存,廖锡文,韦瑞丽,黄伟,龚艺贞,马辉△
1 广西医科大学第一附属医院结直肠肛门外科 广西南宁 530021
2 广西医科大学第一附属医院肝胆外科 广西南宁 530021
直肠癌是消化道最常见的恶性肿瘤之一,其发病机制被认为与环境、遗传因素双重作用驱动有关。对直肠癌进行全基因组测序有助于提高对直肠癌发病分子机制的认识,了解其遗传学上的变异,以期对直肠癌的诊断、预防、监测和靶向治疗上带来一定帮助。随着高通量测序技术的发展,多种癌症在基因组层面上的变异被逐渐揭露,并绘制成癌症相关的全基因组图谱。越来越多的高通量测序相关的基因组研究揭示了包括肿瘤在内的遗传相关疾病的基因组变异和遗传机制,同时,这些高通量测序数据被开放获取分享在开源数据库中供其他研究者使用。癌症基因组图谱(The Cancer Genome Atlas,TCGA)是美国癌症研究所和美国人类基因组研究所合作开展的一个癌症基因组项目,该项目包含了包括33种癌症、超过一万个癌症样本的全基因组多组学高通量测序数据,并分享在其门户网站中供全球研究者无限制的开放获取和利用[1]。本研究的目的是采用TCGA的直肠癌全基因组RNA测序数据和基因集富集分析(Gene Set Enrichment Analysis,GSEA)方法,通过比较癌组织与癌旁组织的全基因组数据集探索直肠癌相关发病机制,现报告如下。
1 材料和方法
1.1 伦理审批
由于本研究的所有数据均来源于TCGA门户网站(https://portal.gdc.cancer.gov),而纳入TCGA的所有样本均获得患者的知情同意并通过相应的伦理审批,且TCGA所产生的数据已开源发布在其门户网站上,并无限制允许研究者对这些数据进行二次挖掘和使用(https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga/using-tcga/citingtcga)。同时,本研究未涉及人体或动物相关的实验操作,因此,本研究无需额外的伦理委员会批准。
1.2 数据下载与预处理
本研究纳入的直肠癌全基因组RNA测序数据集从TCGA门户网站下载,共获得177个直肠癌患者样本的Level 3 RNA测序数据集(Workflow Type:HTSeq-Counts),其中癌旁正常组织10个,癌组织167个[2]。原始的RNA测序数据集经DESeq软件包在R平台中进行归一化[3],归一化后共获得包含17 541个编码基因表达谱数据集用于后续分析。
1.3 基因集富集分析
基因集富集分析的软件(gsea2-2.2.3.jar)和对照基因集从GSEA官网下载(http://software.broadinstitute.org/gsea/index.jsp)[4-5],以癌组织与癌旁正常组织作为表型分组依据。从GSEA网站的Molecular Signatures Database(MSigDB)中选取c2(Curated gene sets:c2.all.v6.2.symbols.gmt)和c5 [Gene ontology(GO)gene sets:c5.all.v6.2.symbols.gmt]作为对照基因集用于直肠癌发病机制的探索[6]。GSEA设置如下:Expression dataset选择输入归一化后的直肠癌全基因组RNA测序数据集;Gene sets database分别选取c2和c5对照基因集;Number of permutations设为1000;Phenotype labels设为癌vs.癌旁;Collapse dataset to gene symbols选False;Permutation type选Phenotype。
1.4 统计学分析
GSEA分析中的FDR的计算根据Benjamini-Hochberg程序进行多次计算得到[7]。GSEA分析结果满足错误发现率(false discovery rate,FDR)<0.25,|Normalized Enrichment Score(NES)|>1 和 NominalPvalue<0.05视为差异有统计学意义。
2 结果
2.1 癌组织与癌旁正常组织在c2富集分析结果的比较
在c2富集分析结果中,在癌组织表型分组中获得170条差异具有统计学意义的基因集,在癌旁正常组织表型中则获得341条(图1A)。在癌组织表型的c2富集分析结果提示直肠癌发病可能参与以下通路机制:细胞周期通路(cell cycle,图1B),核转录因子κB信号通路【nuclear factor-kappa B(NF-κB)signaling pathway,图1C】,DNA修复(DNA repair,图1D),DNA复制(DNA replication,图1E),同时癌与癌旁之间比较可以富集到既往结直肠癌研究中上调的基因集 (colon and rectal cancer up,http://software.broadinstitute.org/gsea/msigdb/cards/GRADE_CO -LON_AND_RECTAL_CANCER_UP,图1F)。
2.2 癌组织与癌旁正常组织在c5富集分析结果的比较
在c5富集分析结果中,在癌组织表型分组中获得151条差异具有统计学意义的基因集,在癌旁正常组织表型中则获得710条(图2A)。在癌组织表型的c5富集分析结果提示直肠癌发病可能参与以下生物学功能:DNA复制(DNA replication,图2B),有丝分裂细胞周期检查点(mitotic cell cycle checkpoint,图 2C),DNA 修复 (DNA repair,图 2D),DNA损伤检查点(DNA damage checkpoint,图2E),错配修复(mismatch repair,图2F)。
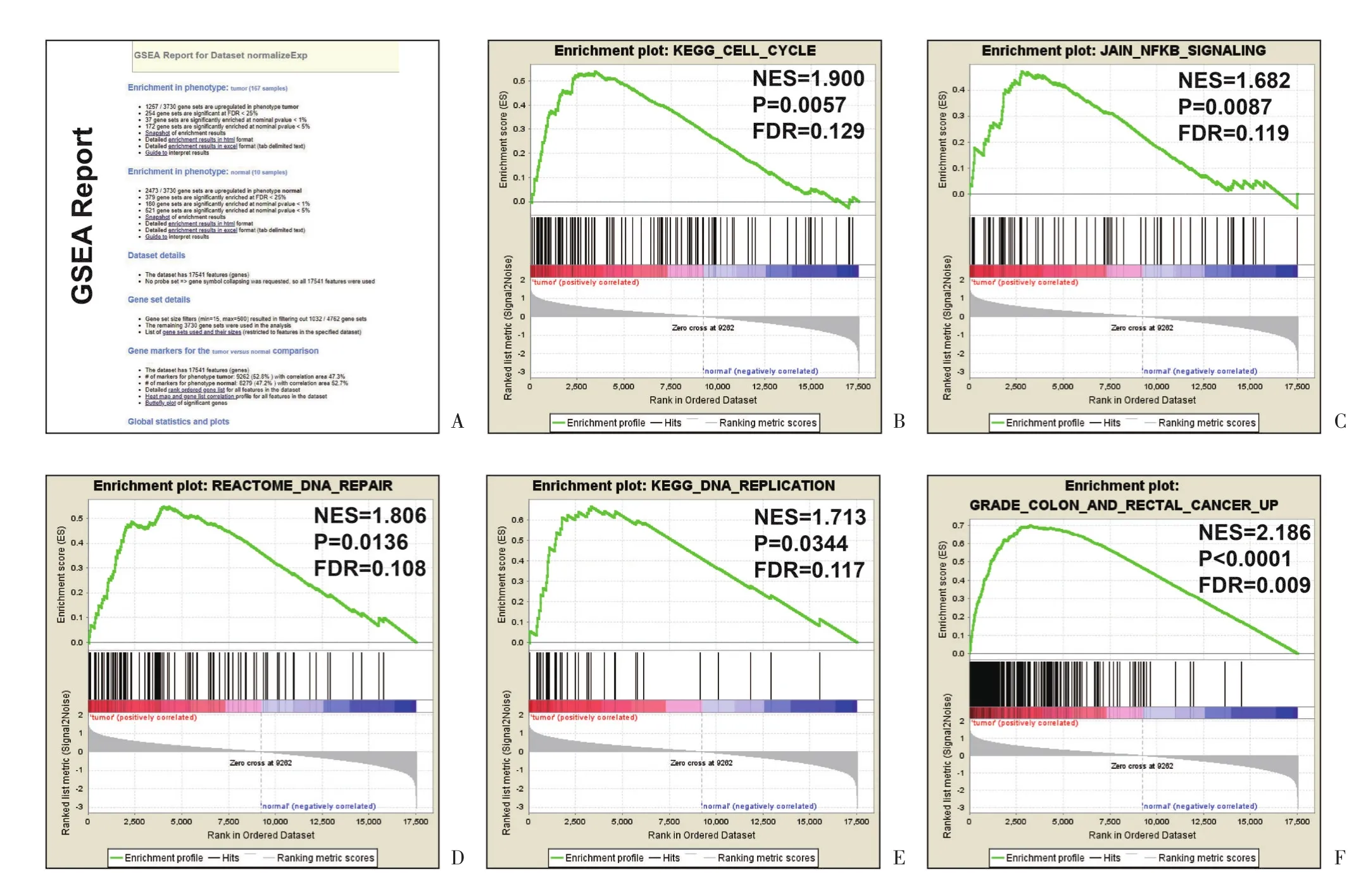
图1 基于癌组织与癌旁正常组织全基因组表达谱数据集以c2作为对照基因集的GSEA富集分析结果
3 讨论
传统的富集分析是通过对比不同分组或表型之间的全基因组数据集,通过设定指定的阈值进行差异表达基因的筛选,随后通过利用差异表达基因通过基因本体论(gene ontology,GO)术语和京都基因与基因组百科全书(Kyoto encyclopedia of genes and genomes,KEGG)等基因功能注释软件进行基因集功能富集。但是这一筛选方法存在一定的缺陷,阈值的设定具有一定主观性和局限性,同时可能令接近设定阈值但具有重要意义的基因被排除在富集分析之外。而GSEA富集分析方法则避免了传统富集分析方法设定阈值的问题,将全基因组数据均纳入富集分析。
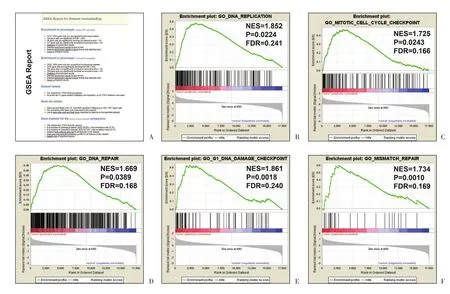
图2 基于癌组织与癌旁组织全基因组表达谱数据集以c5作为对照基因集的GSEA富集分析结果
本研究GSEA富集分析发现细胞周期、DNA复制这两个维持细胞增殖的两个基本生物学过程在直肠癌组织中被显著富集到,提示在直肠癌组织中细胞周期和DNA复制这两个细胞基本状态的生物学过程失调,其与肿瘤的发生有关,而抑制该两个生物学过程则可能抑制肿瘤细胞的增殖。恶性肿瘤的发生与肿瘤基因组异常有关,譬如DNA损伤和修复过程失调,直肠癌的发生同样被认为存在相同的机制[8]。本研究GSEA分析发现直肠癌组织中DNA损伤检查和DNA修复生物学过程和通路被显著富集到,提示肿瘤组织的DNA损伤和修复异常,与肿瘤发生有一定联系。Reilly等[9]的研究也发现DNA损伤反应和修复相关的基因可驱动结直肠癌的发生,同时他们的研究还提示靶向替代DNA修复机制的治疗可以改善结直肠癌患者的预后。此外,有研究还表明错配修复相关基因可预测直肠癌术前放疗的反应率[10-11]。NF-κB信号通路在直肠癌预后预测和放化疗的疗效反应中起重要作用[12-14]。Voboril等[12]研究发现总高水平的NF-κB/p65亚基可能与更具侵袭性的肿瘤特征,更高的肿瘤转移潜能和缩短的总生存期相关,但它与对(化疗)放疗的抵抗无关。Dzhugashvili等[13]研究发现坐落于NF-κB通路中的NFKB1基因的rs28362491的基因型与直肠癌化疗后的病理反应相关。在Berardi等[14]的研究中,通过免疫组化检测NF-κB的表达水平,发现直肠癌癌组织中NF-κB免疫组化表达阳性的患者总体生存时间显著较阴性患者短,且中位进展时间明显缩短。O’Neil等[15]发现NF-κB p50高表达的直肠癌患者其预后较差,通过关联分析推测出NF-κB在直肠癌中起关键作用可能是其参与肿瘤的转移,而不是对放化疗的抵抗。本研究通过全基因组GSEA分析也发现NF-κB信号通路在直肠癌肿瘤的发生中扮演一定的角色。本研究具有一定的局限性需要说明。第一,纳入本研究的癌旁样本量较少;第二,本研究数据来源于单一数据库,缺乏基础实验验证,本研究产生的结果还有待进一步实验确认。尽管存在上述局限性,但本研究的结果仍可为直肠癌发病机制的研究提供一定的理论依据。
综上所述,本研究利用TCGA直肠癌队列的癌组织与癌旁正常组织全基因组RNA测序数据集进行GSEA富集分析发现,直肠癌的发病机制可能参与调控细胞周期、DNA修复、DNA复制等生物学过程和NF-κB等信号通路,提示直肠癌发病机制可能涉及细胞基本状态的调控。
