
串珠藻目植物psaA基因的适应性进化及共进化分析
2019-08-22 01:27:32巩超彦南芳茹冯佳吕俊平刘琪刘旭东谢树莲
山西大学学报(自然科学版) 2019年3期
巩超彦,南芳茹,冯佳,吕俊平,刘琪,刘旭东,谢树莲
(山西大学 生命科学学院,山西 太原 030006)
串珠藻目Batrachospermales是淡水红藻中最重要的代表类群,零星式分布于流动的冷泉溪中,种群多小而少。一般认为包括3科,14属[1-3]。
psaA是植物叶绿体进行光调节的重要基因,位于叶绿体基因组的大单拷贝区[4]。它能编码一种类囊体膜蛋白,即光系统Ⅰ反应中心跨膜复合物的中心蛋白A,又称P700脱辅基蛋白A,该蛋白与psaB基因编码的蛋白相互结合,共同作为光系统Ⅰ跨膜复合物的中心蛋白,承担重要的生物学功能,构成植物光系统Ⅰ的中心架构。
适应是生物进化的核心,物种的演化历程即不断适应其生存环境的过程。在此过程中,物种的遗传信息发生了改变,其中的一些特殊的改变经过自然选择被固定了下来,即基因的适应性进化[5]。同时,分析基因的适应性进化还有助于我们了解基因变异和蛋白质结构与功能的改变及物种的进化史[6]。目前用于进化研究的模型有机理式模型、经验式模型和机理-经验式模型[7-10],而且这些模型也在应用中得到了不断完善[11-17]。
氨基酸位点的共进化是在蛋白质内部和蛋白质间普遍存在的现象[18]。共进化位点的关联特性可能源于蛋白自身结构、功能特性、编码核苷酸构成基因组特性等。如果其中某一位点发生了适应性进化,与此相关的位点通过共进化的方式进行补偿突变,这样不仅可获得有利于增加适应性的新性状,同时也维持了其固有的功能[19]。识别共进化氨基酸位点对了解蛋白质氨基酸位点间可能的相互作用和蛋白适应性进化的复杂机制具有重要意义。
虽然一般认为序列高度保守是叶绿体基因组的特性之一[10],但对于起源较早、演化历史更长的低等植物,可能会发生更多的变化。为了进一步了解串珠藻目植物的演化历史,了解这一特殊的淡水红藻类群究竟如何适应特殊生存环境,相关叶绿体基因是否在其演化历程中发生了适应性进化,从而对其适应生存环境产生影响[20],本研究对其叶绿体基因psaA进行了适应性进化及共进化分析,为了解串珠藻目植物适应生存环境的分子进化机制,了解相关蛋白质内部氨基酸分子之间的相互关系及对该类群适应性进化的影响提供科学依据。
1 材料与方法
1.1 序列数据
本文所用数据包括作者采集样本测得和从GenBank收集的串珠藻目及近缘类群psaA基因42条 (表1)。其中,作者采集的7株样本包括Batrachospermumgelatinosum(采于山西宁武县汾河源头,水温11℃,生于溪流中石壁上),B.hondongense(采于山西洪洞广胜寺泉,水温14℃,pH 7.5,生于急流中石块上),B.longipedicellatum(采于江苏徐州拔剑泉,水温16℃,pH7.5,生于石灰质岩石上),Kumanoacurvata(采于湖北当阳珍珠泉),K.intorta(采于山西临汾龙祠泉,水温14℃,生于泉眼边岩石上),Sheathiaarcuata(山西太原晋祠泉,水温12℃,pH 7,生于小溪流中岩石上)和Thoreahispida(山西平定娘子关泉,水温15.5℃,pH 6.5,生于泉水中树枝上)。
通过软件Clustal X[21]分析,得到由231个密码子组成的序列数据。
1.2 系统发育树构建
利用MEGA5.0分析序列特征,运行Modeltest软件,筛选核苷酸最优进化模型。采用最大似然法(ML)运行PhyML3.0构建系统发育树[14]。
1.3 适应性进化分析
利用基于psaA基因构建的ML树,运行PAML4.8软件包中Codeml程序中的分支模型(用于提示各分支的选择压力,分别运行单比率模型、自由比率模型和二比率模型并进行LRT检验以确定哪个模型更可靠)、位点模型(用于检验基因是否存在经受正选择(ω>1)和负选择(ω<1)的位点,分别运行M1a和M2a,M0和M3,M7和M8三对模型,前者是后者的零假设,对3对模型分别进行LRT检验)和分支-位点模型进行分析(用于检验各个指定分支中是否存在正选择位点)。
1.4 三维结构建模
以B.gelatinosum的psaA(KM055293)序列为参考,翻译为氨基酸序列,提交瑞士生物信息研究所(https:∥www.expasy.org/),基于同源建模原理预测其蛋白的三维结构。
1.5 共进化分析
运用基于Pearson相关系数法[22]、参数检验法[23]和互信息法[24]等的CAPS(Coevolution Analysis using Protein Sequences)软件[25]分析蛋白质量部的共进化关系。
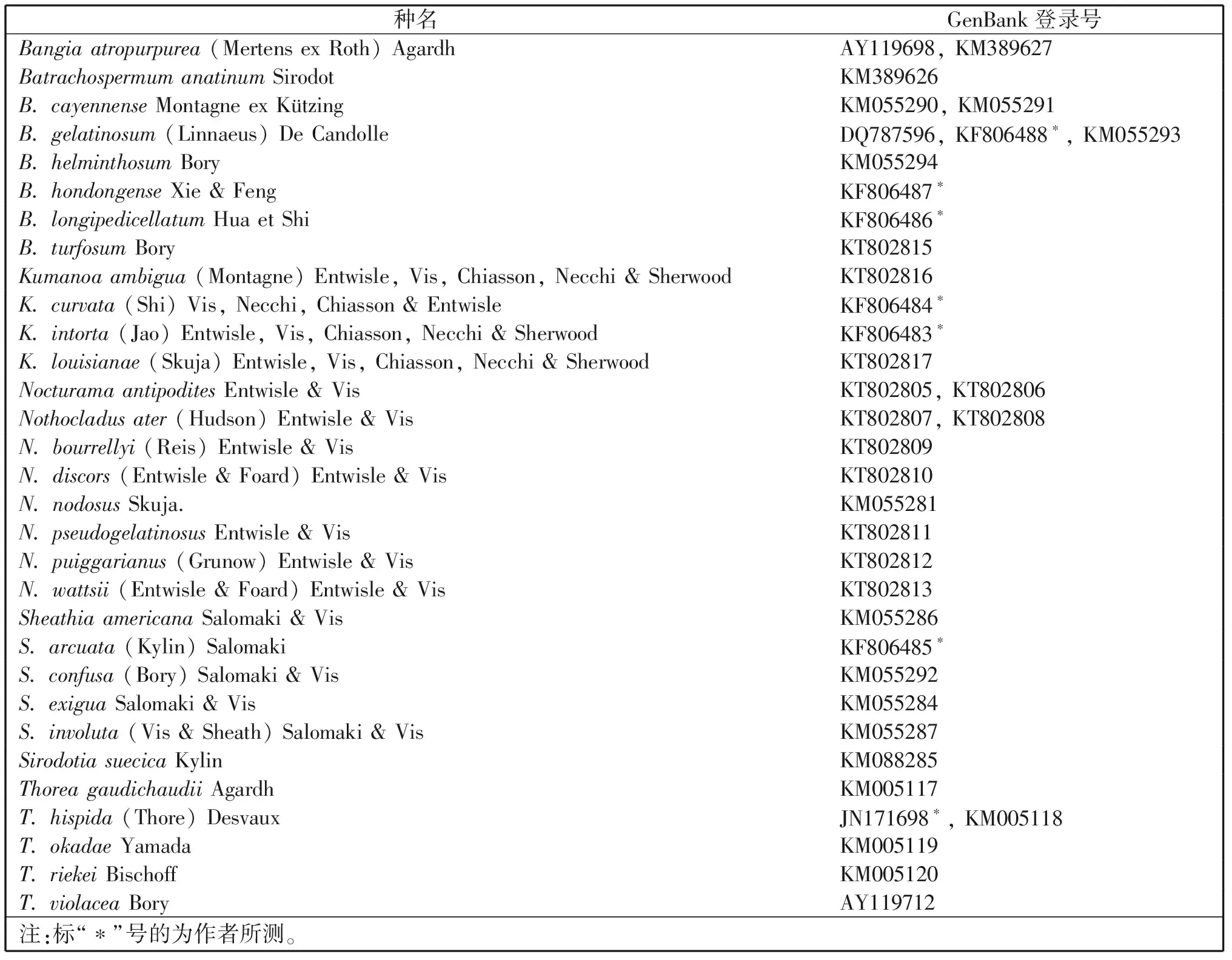
表1 种类及psaA基因的GenBank登录号
2 结果与分析
2.1 系统发育分析
基于选取的psaA序列,得到核苷酸最优进化模型(表2),并基于此构建了系统发育树(图1),外类群为Galdieriamaxima和Cyanidiumcaldarium。
从图1可以看出,内类群聚集为9个分支,包括Nothocladus属、Nocturama属、Sirodotia属、Kumanoa属、Sheathia属、Bangia属、Thorea属、Batrachospermumgelatinosum和其余几个种分别位于不同分支。
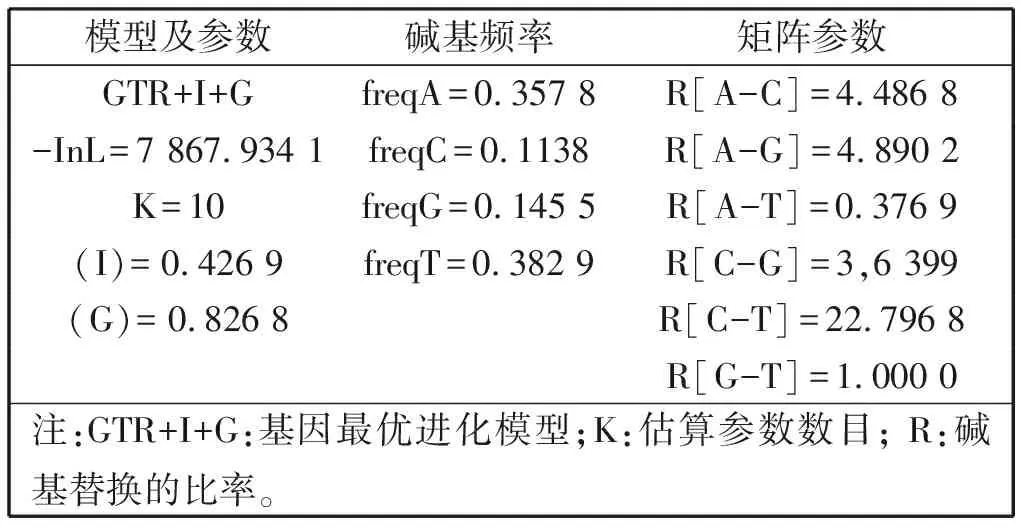
表2 psaA基因最优进化模型参数
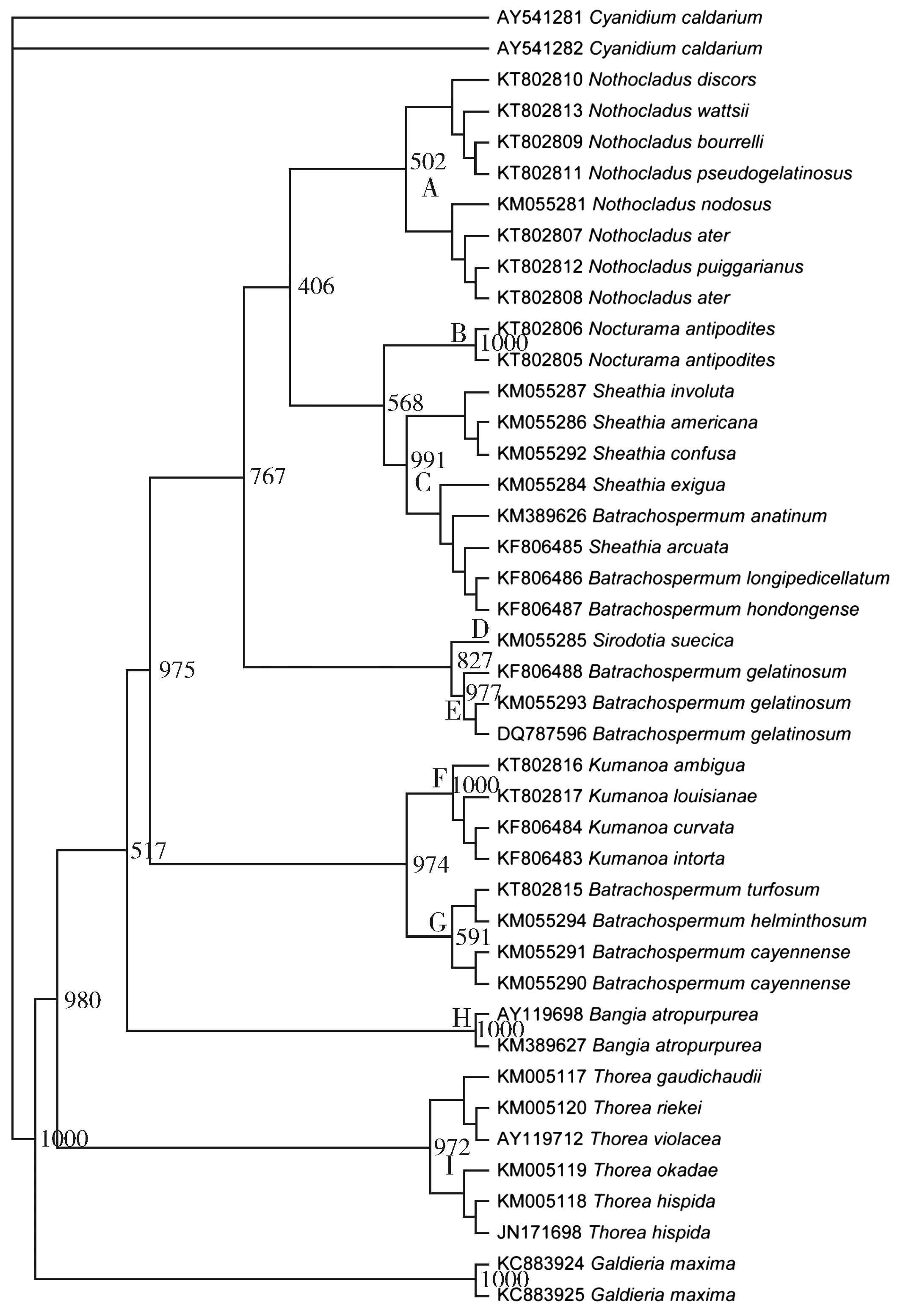
注:节点处的数字代表最大似然法歩靴值,A、B、C、D、E、F、G、H、I代表选定的分支。Fig.1 Phylogenetic tree established based on the psaA gene sequence图1 基于psaA基因序列构建的系统发育树
2.2 正选择位点的鉴定分析
表3和表4是位点选择的结果。可以看出,分支模型中没有正选择位点被检出,说明psaA基因受到强烈的负选择。分支-位点模型中,分支CSheathia属的186F、分支EB.gelatinosum的100A、分支HB.atropurpurea的90C*和199A、分支IThorea属的99A、132A和179M被鉴定为正选择位点。但是LRT检验均显示上述结果不可靠。另外,分支A、B、D、F、G的备择假设经LRT检验均被拒绝。
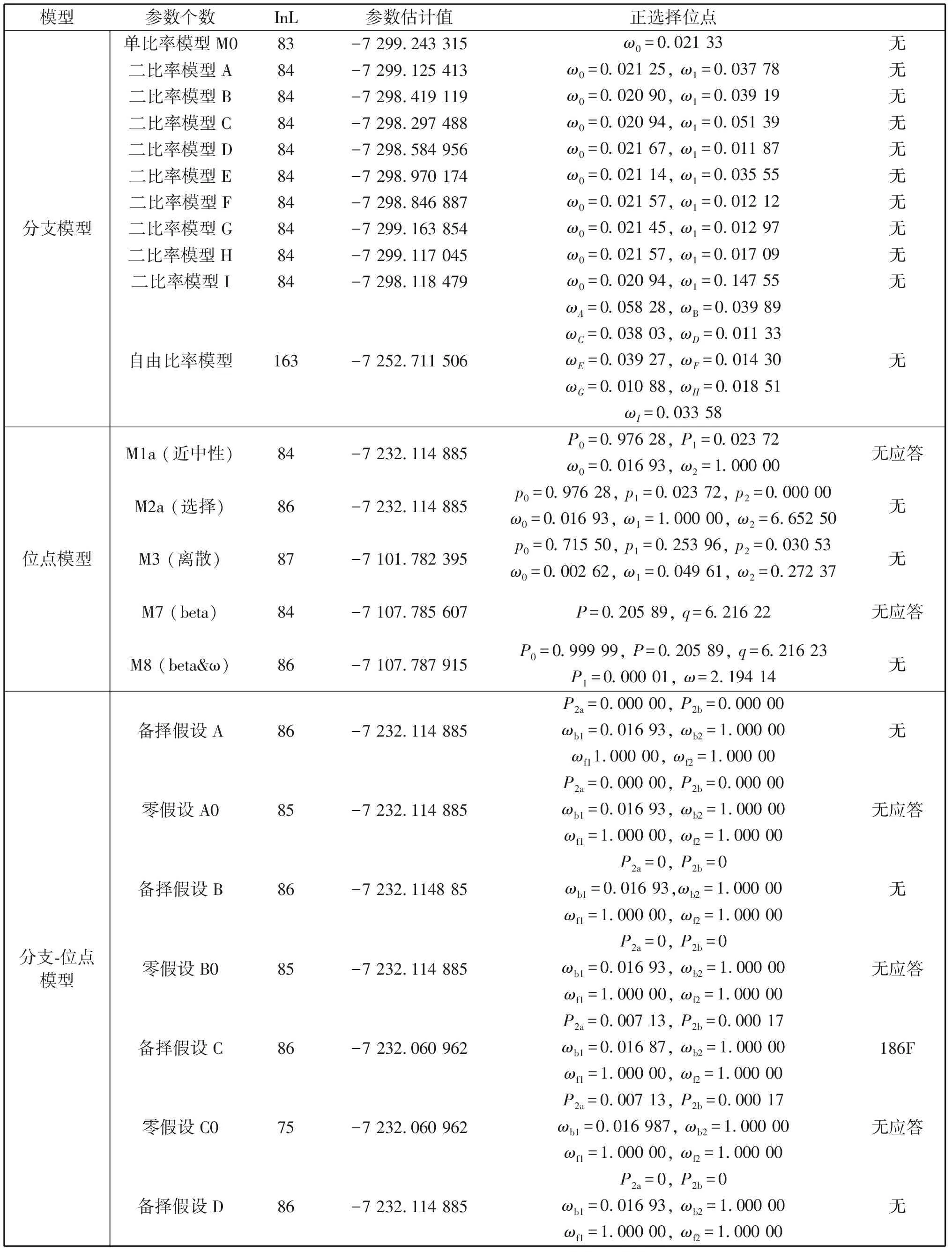
表3 模型参数估计值和对数似然值
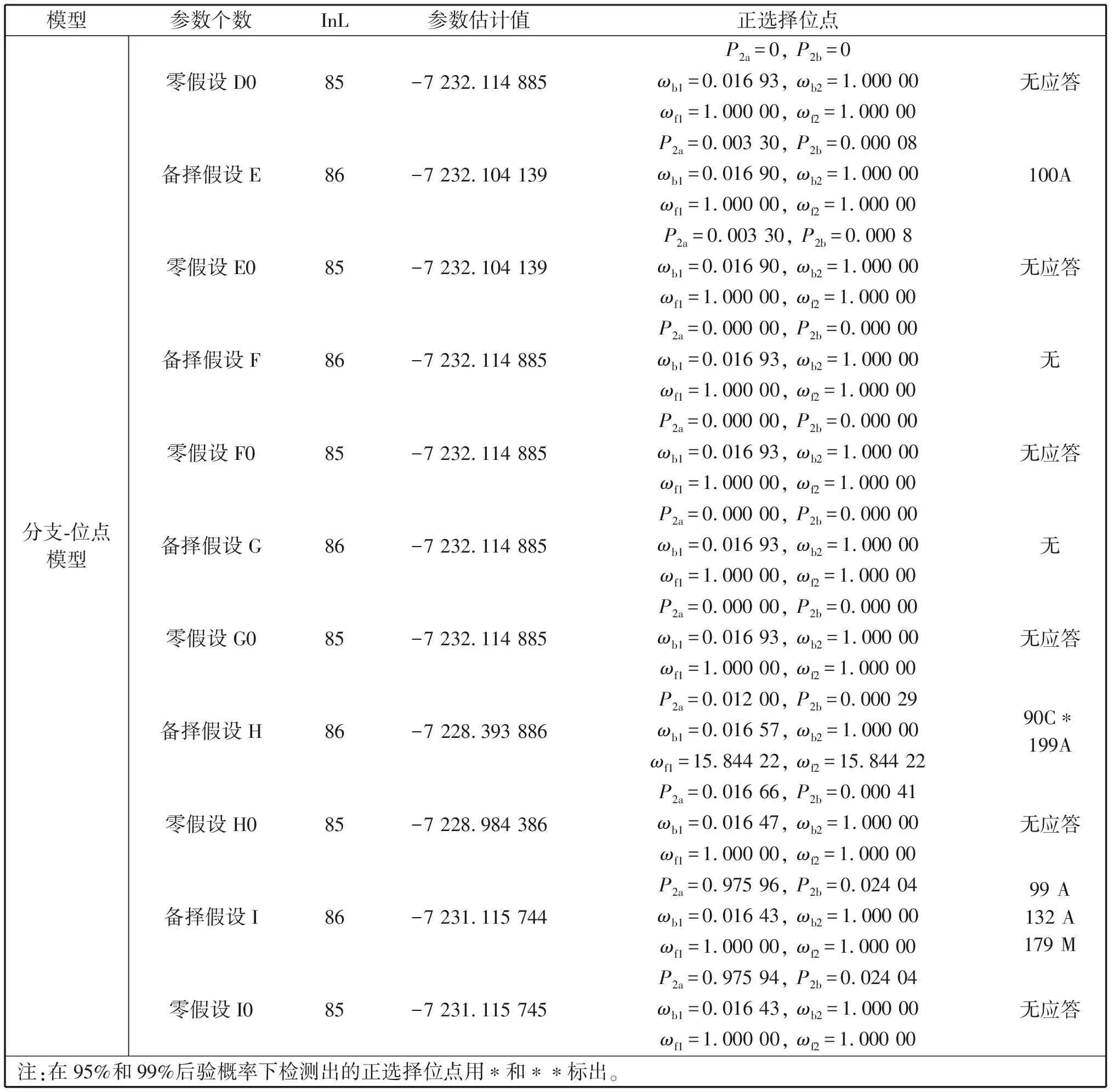
续表3 模型参数估计值和对数似然值
2.3 psaA基因的参考三维结构
以B.gelatinosum登录号KM055293为参考序列,对PDB数据库进行Blast搜索模板,得到一株Thermosynechococcuselongatus(strain BP-1)的三维结构(PDB ID:4FE1)[26],相似度81.99%,一株Synechocystissp. (PCC 6803)的三维结构(PDB ID:4KT0)[27],相似度81.8%,均符合同源建模的可靠性要求。基于前者构建出了psaA蛋白的三维结构(图2)。但由于前者氨基酸序列同分析序列比对有缺失,因此选取后者用于后续共进化分析。
2.4 共进化位点鉴定分析
通过实验序列和已解析的psaA蛋白质三维结构(PDB ID:4KT0)的比对确定对应氨基酸的具体位置(图3),基于详细的解析数据和氨基酸对的相关系数统计出的共进化组(对)5组(20对)(表5)。序列中所有共进化氨基酸对的平均距离27.808 6 Å,标准差12.325 2。基于氨基酸疏水相关性值统计出的共进化组(对)5组(12对)(表6)。基于氨基酸分子量相关值统计出的共进化组(对)5组(15对)(表7)。几对相关系数较高的共进化位点在构建出的参考三维结构中的位置见图4。
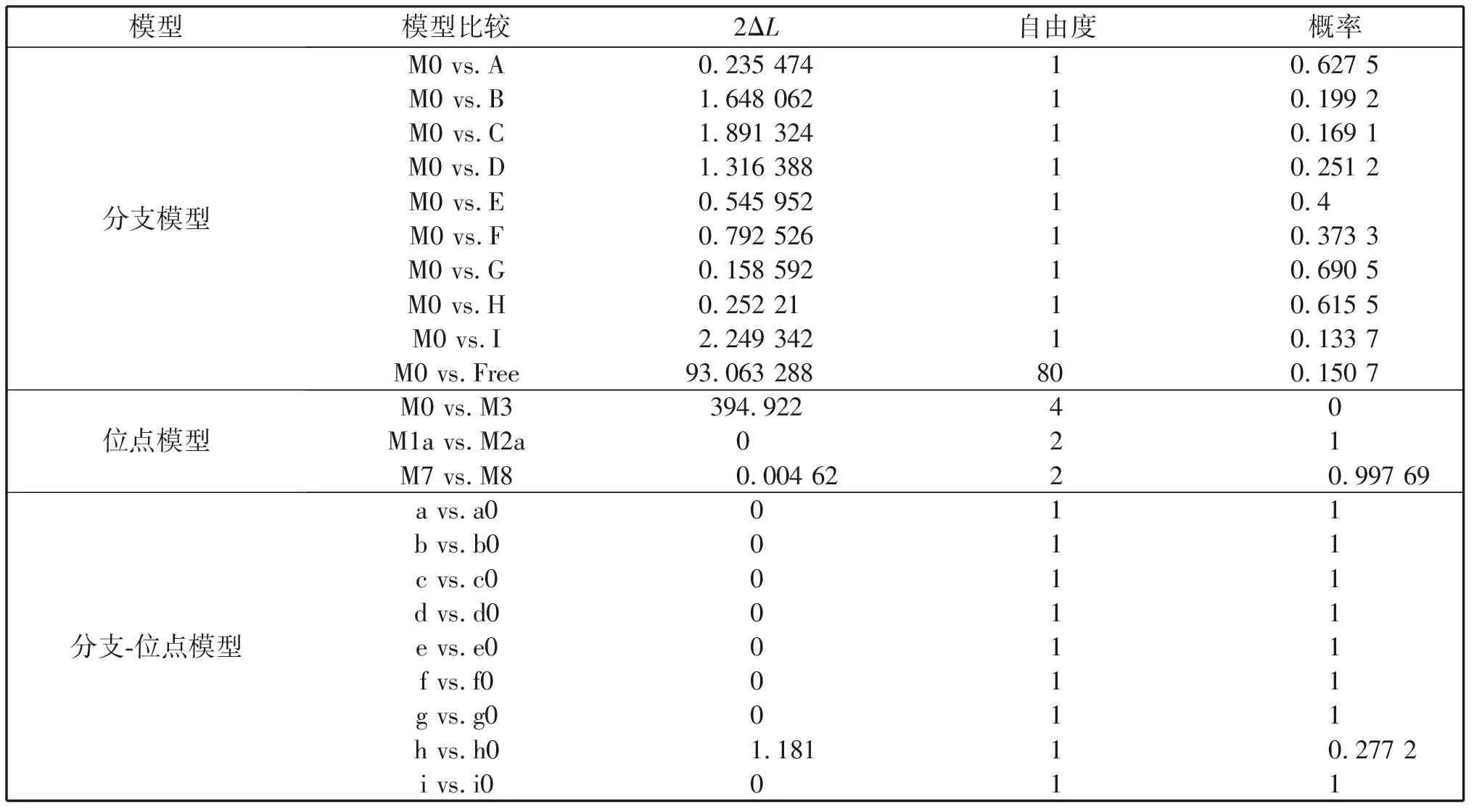
表4 LRT检验统计量

注:正选择位点在图中用白色圆圈标出Fig.2 Reference three-dimensional structure of D1 protein of Batrachospermum gelatinosum (DQ787636)图2 Batrachospermum gelatinosum(DQ787636)D1蛋白参考三维结构
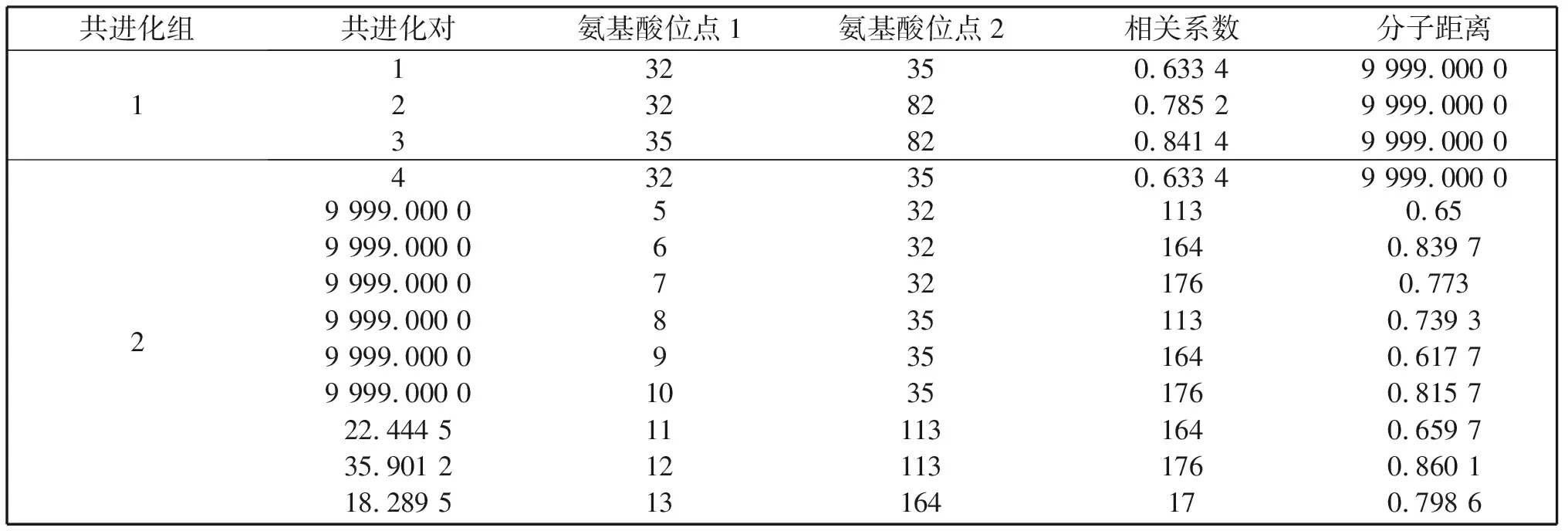
共进化组共进化对氨基酸位点1氨基酸位点2相关系数分子距离 1132350.633 49 999.000 0232820.785 29 999.000 0335820.841 49 999.000 0 2432350.633 49 999.000 05321130.659 999.000 06321640.839 79 999.000 07321760.7739 999.000 08351130.739 39 999.000 09351640.617 79 999.000 0 10351760.815 79 999.000 0 111131640.659 722.444 5121131760.860 135.901 213164170.798 618.289 5
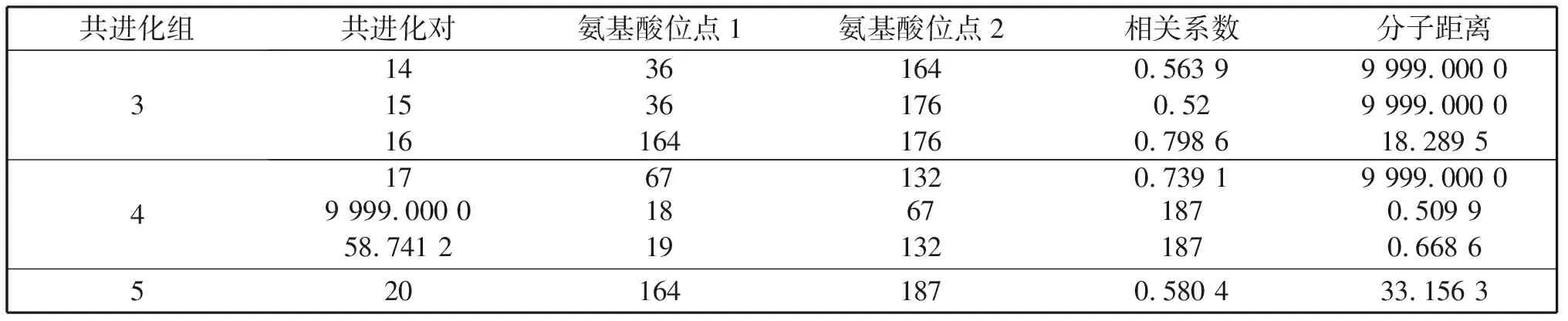
续表5 基于氨基酸对相关系数统计出的psaA蛋白的共进化组(对)
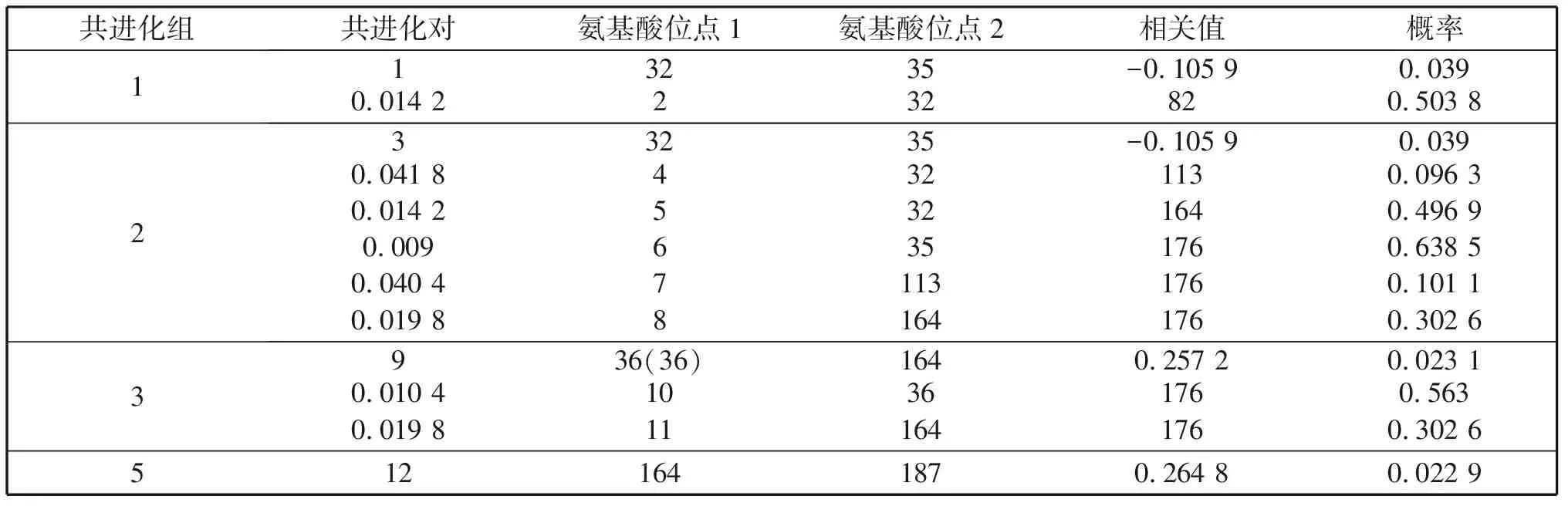
表6 基于氨基酸疏水性相关性值统计出的psaA蛋白的共进化组(对)
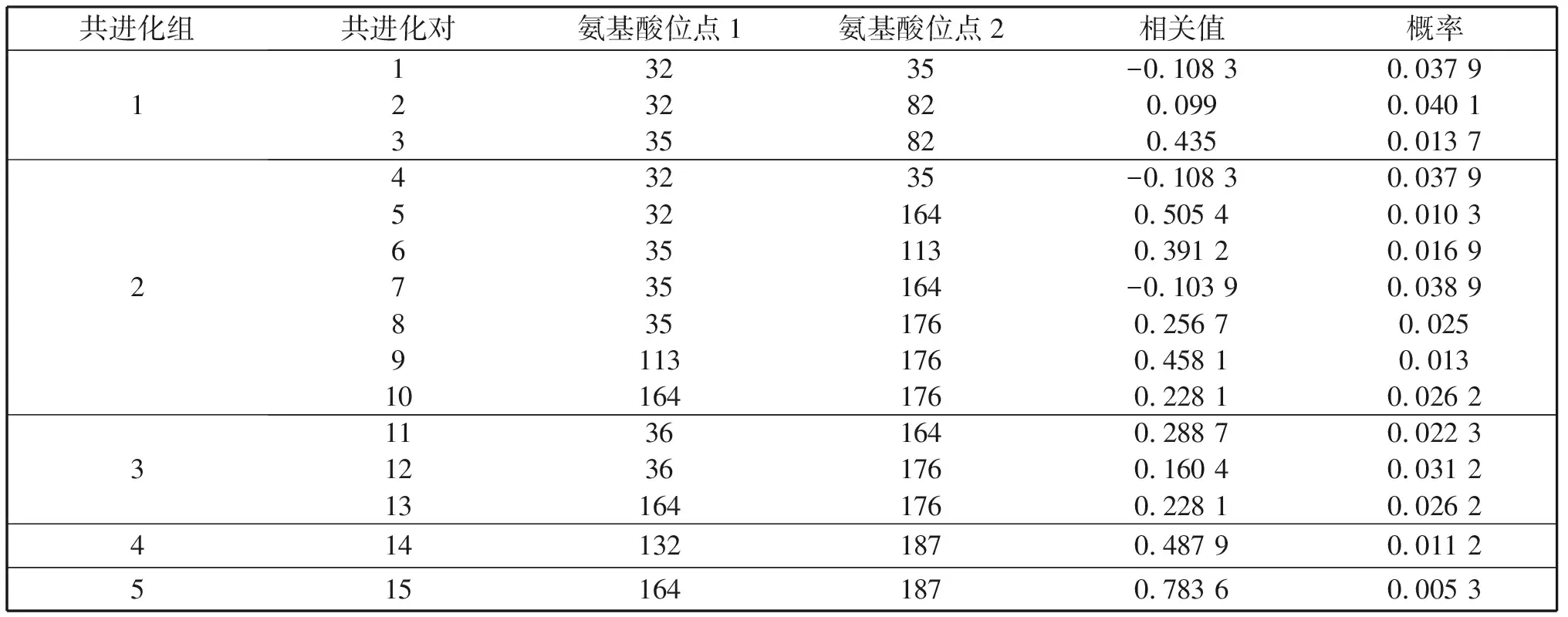
表7 基于氨基酸分子量统计出的psaA蛋白的共进化组(对)

Fig.3 Alignment of the amino acid sequence of psaA protein图3 psaA蛋白质氨基酸序列对位排列
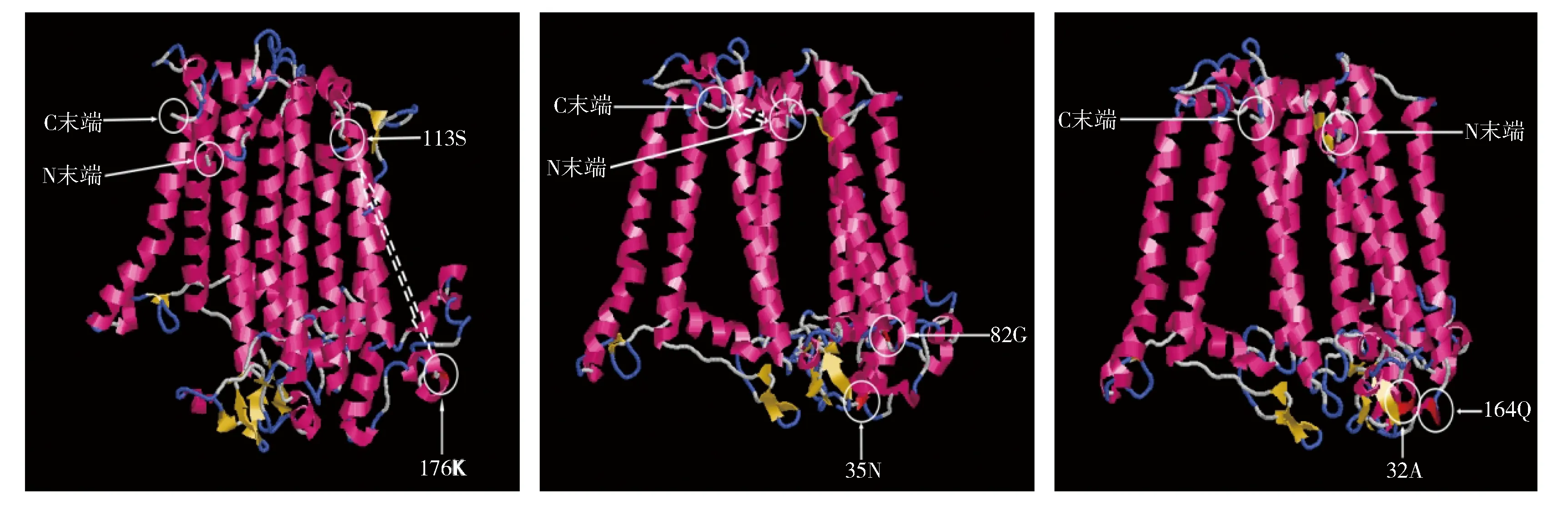
注:共进化位点在图中用白色圆圈标出Fig.4 Spatial location of three pairs co-evolution sites图4 3对共进化位点的空间位置
3 讨论
一般认为序列高度保守是叶绿体基因组的特性之一。一个蛋白为了维持其特定的功能,其结构具有一定的保守性,因而会处于负选择作用之下。本文对串珠藻目psaA基因的研究结果显示,各分支均没有检测出有统计学意义的正选择位点,可以推断串珠藻目植物叶绿体的psaA基因处于强烈的负选择之下,这与该基因的保守性有很大的关系。Smart等[28]发现psaA或psaB基因的失活都将导致PSⅠ复合物在类囊体中缺失,这表明psaA或者psaB不能单独形成二聚体,而psaA-psaB异二聚体的存在为整个PSⅠ复合物组装所必需,可见psaA基因编码的PSⅠ中心蛋白A在光合作用中的重要作用。陈晓霞等[10]研究也指出,有关叶绿体基因发生适应性进化的报道尚不多见,可能的原因主要是叶绿体基因的替换率低,较少发生突变,或者叶绿体基因很少发生重复,缺乏产生新基因的来源。郁飞等[29]研究指出,高等植物、藻类和蓝细菌所有已知的编码PSⅠ结构蛋白的基因都已经被克隆,一级结构相当保守。此外,关于psaA基因的同源性研究也提示了该基因的高度保守性。Cantrell等[30]研究指出蓝藻psaA基因序列同已测序的高等植物有超过95%的同源性,翻译产物和高等植物的同基因产物也有76%~81%的一致性。这种高度的同源性提示,不仅是PSⅠ复合物的结构保守,其功能也相当保守。施定基等[31]比较了蓝藻叶绿体PSⅠ的蛋白基因同若干高等植物的同源性,结果在74.60%~80.19%之间,而且物种间psaA基因的同源性与它们的进化程度相关,进化地位越相近,同源性越高。
基于氨基酸对的相关系数统计出的共进化组(对)之间相互关联非常紧密,如果其中某一位点发生了适应性进化,与此相关的位点通过共进化的方式进行补偿突变,以维持蛋白质特定的功能。基于氨基酸疏水性相关性值统计出的共进化组(对)揭示它们的共同作用对保持蛋白质疏水性具有重要作用。基于氨基酸分子量相关性值统计出的共进化组(对)则提示它们同蛋白质分子量大小显著相关。本文分析得到若干对串珠藻目植物psaA基因内部不同位置的共进化组(对),从另一个角度分析了蛋白质内部氨基酸之间的相互关系,有助于研究蛋白质的功能结构特征及其进化历程,为研究有关蛋白结构功能和进化提供了优先考虑的位点。
随着分子生物学的发展,psaA基因作为标志分子在藻类植物分类系统发育研究中的应用也值得关注。一般来说,叶绿体基因组具有基因组较小、在进化过程中很少发生重排、进化速率保守、包含大量的DNA成分可提供足够数量的系统发育信息、编码区和非编码区序列进化速率相差较大等特征[32-33],因此,非常适用于不同分类水平的系统发育分析研究。一般认为,功能蛋白编码基因受其功能限制,进化速率慢,适于属间及以上分类单元群体的关系研究,而非编码区基因不参与蛋白质的合成,进化速率相对较快,多样性较高,适合于种内及种间的群体关系研究[34-35]。本研究结果显示,psaA基因在串珠藻目中是很保守的,是属及以上分类单元群体的系统发育关系研究的重要分子标记。
生物体演化的历程和分子机制是极其复杂的,因此,要解释清楚为何淡水红藻分布区狭窄、对生境的要求很特殊,需要继续对更多的基因展开深入研究,进一步深化对叶绿体基因为适应环境而发生的分子水平的适应性进化机制的认识。
猜你喜欢
农业科技通讯(2023年1期)2023-02-12 07:07:54
学生天地(2019年28期)2019-08-25 08:50:54
中国外汇(2019年23期)2019-05-25 07:06:20
数学物理学报(2018年1期)2018-03-26 08:16:36
北京航空航天大学学报(2016年9期)2016-11-16 02:02:33
广西林业科学(2016年1期)2016-03-20 05:33:01
现代检验医学杂志(2015年6期)2015-02-06 01:43:55
山西大同大学学报(自然科学版)(2014年3期)2014-01-23 01:56:30
食品科学(2013年6期)2013-03-11 18:20:13
疯狂英语·口语版(2013年1期)2013-01-31 09:23:26
