
Peutz-Jeghers综合征研究进展
2019-08-17 07:02陈志祥孟立娜
胃肠病学 2019年6期
陈志祥 孟立娜
浙江中医药大学附属第一医院消化科(310006)
Peutz-Jeghers综合征(PJS)是一种罕见的常染色体显性遗传疾病,其发病率约1/20万[1],其中50%的患者伴有家族史,临床上可见皮肤黏膜黑斑以及胃肠道多发息肉,且消化道息肉有恶变的风险,恶性肿瘤的发生率可高达20%[2],还会增加其他系统恶性肿瘤的发生率。事实上,如缺乏适当的医疗监督,PJS患者累积癌症的风险高达93%[3]。本病在国内少见,临床医师对其认识尚存不足,故本文就PJS的研究进展作一综述。
一、发病机制
研究表明位于人体19号染色体短臂(19p13.3)的丝氨酸/苏氨酸蛋白激酶11(LKB1/STK11)基因的钝性突变为PJS主要的致病原因。LKB1基因是一种抑癌基因,其表达的LKB1蛋白是一种cAMP依赖的蛋白激酶,通过磷酸化作用调控细胞分化,对维持细胞增殖、凋亡、胚胎、血管发育、细胞极性等细胞功能至关重要。Chen等[4]的研究发现具有致病作用的LKB1基因种系突变是中国家族性PJS患者的常见病因,LKB1基因启动子区甲基化可能导致了胃肠道息肉的癌变。
LKB1基因调控细胞增殖的主要机制是通过p53依赖的途径调控Rb蛋白的磷酸化水平而实现的[5],其中腺苷酸活化蛋白激酶(AMP-activated protein kinase, AMPK)是LKB1主要的下游激酶,具有感知机体能量状态并维持机体能量代谢稳态的作用。George等[6]的研究表明LKB1可能是调节p53依赖的细胞凋亡的信号转导分子之一,其功能缺失可导致肠上皮细胞凋亡缺陷,这种缺陷是PJS患者胃肠道多发良性错构瘤性息肉的始发因素,并最终导致向恶性肿瘤转变。Jiang等[7]的研究发现PJS相关的STK11突变体会产生一种截短蛋白,该截短蛋白会导致p53活性下调,增加癌症发生风险。
LKB1和其同源类似物在不同的模型系统中对细胞极性均起有调节作用。细胞极性丧失是上皮-间质转化(EMT)的标志之一,钟超等[8]的研究表明LKB1可能通过调控EMT从而影响PJS错构瘤的形态改变以及纤维化进程,LKB1缺失的细胞将发生EMT改变,使细胞增殖和迁移能力增强,提示EMT可能成为PJS新的治疗靶点。
目前在PJS患者中已发现了超过400个基因突变[9]。基因的杂合性缺失、移码突变或无义突变使LKB1氨基酸改变或产生截短蛋白,异常的LKB1蛋白激酶不能被正常磷酸化而失去抑瘤功能,进而导致胃肠道息肉形成以及多器官恶变风险增加。Tchekmedyian等[10]发现具有LKB1基因截短突变者可能会发生严重的并发症,手术次数增多且更易患癌症。
二、PJS的临床特征
PJS以广泛分布的皮肤黏膜黑斑和胃肠道多发息肉为临床特征。PJS黏膜黑斑多呈浅棕色,颜色均匀,通常小于5 mm,常见于唇、齿龈、颊黏膜、口、鼻、眼周围,也可分布于小阴唇、龟头等外生殖器。目前尚无PJS患者皮肤黏膜色素沉着恶变的报道[11]。
PJS息肉分布于全消化道,最常见于小肠,其次为结直肠、胃,其中小肠息肉检出常见部位依次为近段小肠(十二指肠和空肠上段)、中段小肠、远段小肠[12]。息肉随着年龄增长而逐渐生长,并可引起急慢性腹痛、肠扭转、肠套叠、肠梗阻、胃肠道出血等严重并发症,其中以小肠套叠最常见,还可出现肛门息肉脱垂[13]、胆管梗阻[14]等少见并发症。研究表明,PJS患者从出生至20多岁时发生肠套叠的风险高达50%,导致肠套叠的息肉直径一般>15 mm,其中95%的肠套叠发生于小肠,多数需接受手术治疗[15]。PJS的表现形式多样,存在体征分离现象,有黑斑而无消化道息肉、无黑斑有消化道息肉、又有黑斑又有消化道多发息肉这三种临床表现[16]。因此,对于临床上仅有消化道多发息肉或仅有黑斑症状的患者,家族史显得格外重要。
多数PJS的病理类型为错构瘤,少数为腺瘤、增生性、炎性、幼年性息肉或多种类型息肉并存,可伴发消化道外其他部位的错构瘤[17]。既往认为PJS息肉恶变途径为错构瘤-腺瘤-腺癌,但随着研究地不断深入,发现PJS的息肉病理多数为腺瘤,这与典型的错构瘤-腺瘤-腺癌恶变途径不符,因此可能存在另一条恶变途径,即腺瘤-腺癌,但尚需大样本量研究进一步证实[17-18]。据报道,PJS患者胃肠道癌变风险升高,累积癌症风险依次为结直肠(39%)、胃(29%)、小肠(13%)[19]。此外,PJS容易并发消化道外恶性肿瘤,如女性生殖细胞瘤、男性睾丸支持细胞瘤、胰腺癌、肺癌、乳腺癌等,其中乳腺癌的发生常伴随胃肠道癌症的发生,患胰腺癌的相对风险为正常人的132倍[20]。因此,临床上应对PJS患者进行及时地监测随访,早发现、早治疗,从而改善预后。
三、PJS的诊断与治疗
1. 诊断方法进展:PJS主要通过家族史、临床特征、影像学以及内镜下胃肠道息肉进行综合诊断。一般影像学检查包括腹部B超、CT、MRI、消化道造影、胃肠镜。对于较难发现的小肠息肉,目前主要通过多层螺旋CT(MSCT)、小肠CT造影(CTE)、磁共振小肠造影(MRE)、胶囊内镜、气囊辅助小肠镜(BAE)等手段进行检查。冯瑞等[21]的研究对29例患者进行MSCT检查,结果显示MSCT对息肉的检出率高达96.6%,对PJS病灶的定位、定性、鉴别诊断和随访具有较高的应用价值和可行性。CTE和MRE检测小息肉(约5 mm)的敏感性高于消化道造影,亦可用于检测>5 mm的息肉。对于5~15 mm 的息肉,MRE和内镜检查的一致性为72.6%,而对于>15 mm的息肉,两种检查方法的符合率为93%[22]。有研究推荐胶囊内镜作为监测小肠息肉的首选检查。胶囊内镜可检出<5 mm的息肉,对于6~10 mm的息肉,胶囊内镜的检出率高于其他影像学检查,但对于>10 mm的息肉,胶囊内镜与其他影像学检查无明显差异[23]。BAE具有安全性高、可重复检查、检查效果确切等优点,可对整个胃肠道进行检查,对PJS息肉的大小、形态、位置的判断准确,可发现套叠的肠管并尝试进行复位[22],并能对内镜下发现的息肉进行治疗。陈燚等[24]将小肠充气螺旋CT三维重建这项新技术用于PJS诊断,结果发现CT经肛充气灌肠对需要临床干预的病灶的检出率为88.2%,对临床小肠进镜方式的选择具有指导价值。
2. 治疗现状:目前对息肉的治疗多采用内镜下切除,必要时行外科手术治疗。手术治疗的适应证包括:①出现肠套叠或肠梗阻等并发症者;②消化道出血者;③内镜下无法摘除的较大的息肉,如>2 cm的小肠息肉或>1.5 cm的结直肠息肉;④小肠息肉广泛密集存在者;⑤怀疑癌变者。手术的主要方式包括肠段切除术、肠段切开息肉摘除术、腹腔镜联合小肠镜息肉切除术、术中内镜联合肠段切除术等。PJS患者通常需接受多次手术治疗,易引起短肠综合征、肠粘连等并发症,影响患者术后的生活质量。对于已确诊的PJS,建议对>1 cm的息肉进行干预治疗。BAE对既往较难处理的小肠息肉有重要的临床意义。李明雪[25]对200例PJS患者行650次BAE检查和治疗,结果显示110例(84.6%)患者未再出现并发症,避免了外科手术,说明BAE镜下切除PJS息肉是一种安全有效的治疗手段。
PJS的治疗药物以选择性环氧合酶-2(cyclooxygenase-2, COX-2)抑制剂和雷帕霉素为代表。洪丽莉等[26]对26例PJS结肠息肉患者采用塞来昔布联合电子结肠镜下高频电凝电切术治疗,结果显示治疗24个月后息肉的平均数目和直径>1 cm的息肉数目明显减少;单纯高频电凝电切治疗组部分PJS息肉出现异变的趋势,联合塞来昔布后该趋势降低。王石林等[27]针对性地对PJS患者采用局部治疗(内镜)+解救治疗(手术)+预防治疗(药物干预)的临床综合治疗模式,即首先对PJS患者进行内镜检查和治疗,对镜下治疗困难者或出现并发症者进行开腹手术,随后再口服塞来昔布治疗6~9个月。初步证明这种临床综合治疗模式对PJS的治疗是积极、安全、有效的,可为PJS的临床规范性治疗提供有益的参考。
段福孝等[28]回顾性分析295例PJS患者发现,PJS可根据黑斑出现年龄和初治年龄分成早发型和迟发型2种亚型:黑斑出现年龄<3岁和(或)初次治疗年龄<14岁的患者定义为早发型,黑斑出现年龄≥3岁和(或)初次治疗年龄≥14岁的患者定义为迟发型。迟发型PJS患者临床症状出现和治疗时间较晚,且病情更重,需更多的内外科干预。因此,建议对迟发型患者在治疗上应更积极、内镜检查随访的次数更频繁。
四、PJS的随访与监测
美国ACG指南建议PJS患者及其一级亲属应进行规律随访:① 8岁后每3年行胃镜检查、全消化道钡餐造影、结肠镜检查。② 30岁后每年行腹部B超检查,了解肝脏、胰腺、肾脏等情况。有条件者每2年行超声内镜检查1次。③男性患者14岁后每年行睾丸检查1次。④女性患者25岁后每年行盆腔检查了解子宫、卵巢情况,每年行乳房检查(详见表1)[29]。关于胃肠道随访,目前各研究结果不一。有研究指出,基础内镜检查应在患者8岁时进行,如发现明显息肉,每3年随访1次,若无息肉则18岁再次行检查,此后每3年1次,或随症状情况缩短周期,并于50岁开始将间隔缩短至1~2年[23]。多重连接依赖性探针扩增试验(multiplex ligation-dependent probe amplification assay, MLPA)[30]以及Sanger测序[31]可检测STK11基因突变,这两项技术为PJS的诊断提供了有效的基因检测技术,尤其是对家族史阴性的PJS患者。
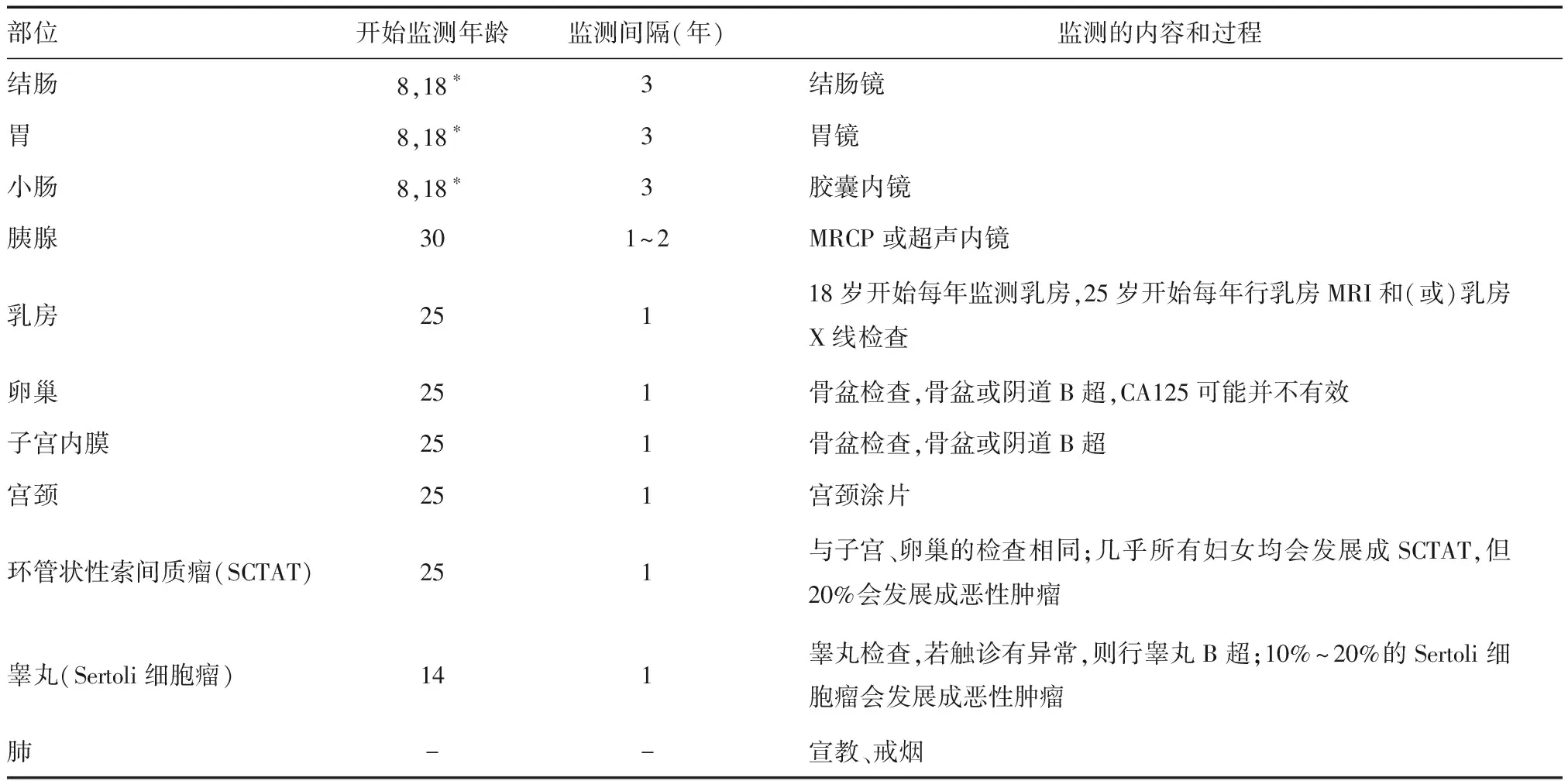
表1 PJS患者的监测[29]
*基础内镜检查在患者8岁时进行,如发现明显息肉,每3年随访1次,若无息肉则18岁再次行检查,此后每3年1次,或随症状情况缩短周期
五、问题和展望
PJS患者早期如何进行治疗以及如何减少息肉复发仍存在许多争议,目前国内外缺少大样本研究证实药物治疗的可靠性,且具体应用缺乏统一标准,PJS肿瘤发生率高,如何进行有效监测和预防,上述问题均是未来研究的重点。随着临床上PJS样本数量的增加以及人类基因组和蛋白组计划的研究进展,PJS这一遗传病有望得到更好的诊治。
猜你喜欢
当代水产(2022年2期)2022-04-26
感染、炎症、修复(2021年1期)2021-07-28
科学导报·学术(2020年47期)2020-11-17
当代水产(2020年4期)2020-06-16
当代水产(2020年4期)2020-06-16
当代水产(2020年3期)2020-06-15
故事会(2019年10期)2019-05-27
食品与健康(2018年3期)2018-03-29
爱你(2016年4期)2016-12-06
养生大世界(2016年3期)2016-04-13
