
不同疾病来源幽门螺杆菌对蒙古沙土鼠胃黏膜损伤影响
2019-08-09 02:23唐义东柳云恩
创伤与急危重病医学 2019年4期
唐义东, 柳云恩, 朴 瑛
1.辽宁中医药大学研究生学院,辽宁沈阳110032;北部战区总医院
2.全军重症(战)创伤救治中心实验室;
3.肿瘤科,辽宁沈阳110016
幽门螺杆菌(Helicobacter pylori,H.pylori)感染人群中少数可发展为慢性胃炎和消化性溃疡,严重者甚至可发展为胃癌[1]。有研究显示,H.pylori不同的致病能力可能与其疾病来源和基因亚型有关[2]。蒙古沙土鼠(Mongolian gerbils,MGs)的腺胃结构和功能与人类相似,自发性胃炎的发生率极低,其可为H.pylori诱导的慢性活动性胃炎、胃癌的发展等研究提供了可用的动物模型[3]。几种天然产物如菜籽油、咖啡酸苯乙酯及蜂胶提取物等对H.pylori诱导的MGs相关性胃炎和胃癌有抑制作用[4]。此外,有研究发现,MGs感染溃疡分离株TN2GF4后会造成明显的炎症、溃疡及腺癌等病理改变,而MGs被溃疡分离株7.13感染14周后,近1/3的MGs发生高分化腺癌,18周后,将近半数的MGs发生高分化腺癌,其子代菌株B128仅能导致MGs发生轻度溃疡和胰腺炎[5-6]。本研究旨在探讨不同疾病来源H.pylori对MGs胃黏膜损伤的影响。现报道如下。
1 材料与方法
1.1 实验动物与材料 雄性SPF级MGs 30只,6周龄,购自浙江省医学科学院实验动物中心。动物饲养在室温(20℃ ±2℃),相对湿度40% ~60%,12 h明暗循环的标准实验室环境。自由摄食、饮水。动物观察及适应环境7 d。不同疾病来源的H.pylori购自中国医科大学肿瘤研究所。
1.2 研究方法
1.2.1 实验动物分组及胃黏膜损伤模型建立 将30只MGs随机分为对照组、胃炎H.pylori组(胃炎株组)和胃癌 H.pylori组(胃癌株组),每组各10只。胃炎株组和胃癌株组MGs禁食禁水12 h后,分别将含不同疾病来源H.pylori的生理盐水1.0 ×108CFU/ml灌胃 l ml,每日1 次,连续3 d,对照组仅灌生理盐水。接种H.pylori后禁食12 h,4 h后自由摄水,连续观察26周。
1.2.2 病理学检查 接种H.pylori后第26周,处死动物。处死前,MGs禁食禁水12 h,乙醚吸入麻醉,对动物胃黏膜进行观察。由前胃、腺胃至十二指肠纵向切成约1.0 cm×0.4 cm条块,然后将组织进行石蜡包埋,用于制作病理组织切片。
1.2.3 免疫印迹试验检测 采用免疫印迹试验检测胃黏膜组织核因子-红系2相关因子2(Nrf2)、Nqo1和Ho-1蛋白表达。将黏膜组织充分碾碎后,在冰上用溶解缓冲液处理45 min,然后将其在95℃下加热5 min,用10%十二烷基硫酸钠-聚丙烯酰胺凝胶电泳,电泳分离等量的蛋白质,移至聚偏二氟乙烯膜。将含有5%牛血清白蛋白的缓冲盐水和TBST阻断膜1 h后,加一抗孵育,4℃过夜,然后用TBST洗涤3次,加二抗培养2 h,使用增强化学发光试剂盒发光。
1.3 统计学方法 采用SPSS 19.0统计学软件对数据进行分析。计量资料以均数±标准差(±s)表示,组间比较采用单因素方差分析。以P<0.05为差异有统计学意义。
2 结果
2.1 不同疾病来源 H.pylori致 MGs胃黏膜病变 建模第26周,对照组胃黏膜未见任何大体改变;胃癌株组80%动物可见胃黏膜糜烂、溃疡,溃疡表现较大,直径约为10 mm,边缘显示不整,伴有黏膜出血,周围黏膜粗糙;胃炎株组40%动物胃黏膜可见糜烂、溃疡,溃疡直径较小,边缘整齐。见图1。

图1 不同疾病来源H.pylori致MGs胃黏膜大体病变
2.2 不同疾病来源H.pylori致MGs胃黏膜组织病理学改变 镜下可见,胃癌株组发生明显的炎症细胞浸润,黏膜糜烂、溃疡,肠黏膜化生等病理改变;而胃炎株组胃黏膜损伤程度较轻,仅见少量炎症细胞浸润,未见明显的黏膜萎缩改变。胃癌株组和胃炎株组导致黏膜炎症的发生率分别80%和40%,黏膜糜烂、溃疡的发生率分别为80%和35%,黏膜发生腺体萎缩、肠化生的概率分别为60%和5%,差异均有统计学意义(P<0.05)。见图2。
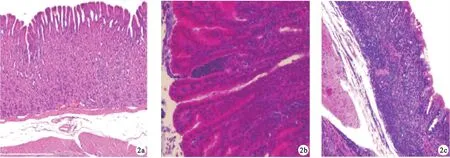
图2 不同疾病来源H.pylori致MGs胃黏膜组织病理学改变
2.3 不同疾病来源H.pylori致MGs胃黏膜Nrf2/Nox4通路蛋白表达 与对照组比较,胃炎株组和胃癌组黏膜Nrf2蛋白的表达增加,Nqo1和Ho-1蛋白的表达降低,差异有统计学意义(P<0.05);与胃炎株组比较,胃癌株组Nrf2蛋白的表达增加,Nqo1和Ho-1蛋白的表达降低,差异均有统计学意义(P<0.05)。见图3。

图3 不同疾病来源H.pylori致MGs胃黏膜Nrf2/Nox4通路蛋白表达
3 讨论
目前,关于H.pylori的持续定植和H.pylori相关胃炎在胃黏膜中的发展尚不清楚,但胃上皮细胞与H.pylori诱导的免疫反应间的相互作用是一个关键的促因。胃上皮细胞能使免疫细胞产生炎症反应[7]。本研究比较了胃癌及胃炎H.pylori分离株感染26周后,MGs胃黏膜的损伤程度。结果发现,第26周,感染胃癌分离株的MGs黏膜及黏膜下层出现明显的淋巴滤泡,糜烂、溃疡、腺体萎缩及肠化生等病变,而感染胃炎分离株则病变程度较轻,仅见轻度的炎症反应等病变。
有研究显示,H.pylori的定植密度与黏膜炎症的严重程度、炎细胞的浸润程度有关,而H.pylori的感染时间和定植密度可调节其致癌作用[8]。H.pylori定植密度及检出率随感染时间的延长而逐渐降低,不同疾病来源的菌株造成MGs损伤程度亦有不同[9]。胃癌分离株TK1402可造成MGs黏膜发生炎症、萎缩及肠化生等病变;而胃溃疡分株TN2GF4菌株虽然也会造成MGs黏膜发生急性胃炎和肠化生等改变,但更容易导致十二指肠炎,不同疾病来源的H.pylori在造成MGs胃黏膜损伤方面比较,差异有统计学意义(P<0.05)[10-12],与本研究结果相似。有研究显示,cagPAI+H.pylori更倾向于诱导MGs黏膜产生溃疡、萎缩、肠化生及增生性病变[13-15];而cagPAI-H.pylori易导致MGs发生糜烂和轻度炎症反应[16-17]。本研究中所采用的两种菌株均为cagPAI+H.pylori菌株,但这两种菌株的致病性仍然存在较大差异。这提示,cagPAI+可能只是决定H.pylori致病能力的因素之一。因此,不同疾病来源的H.pylori对MGs黏膜损伤是一个复杂、多因素、多步骤共同作用的结果,其某些关键基因的突变和缺失可能会在某种程度上降低其致病能力。Nrf2是含转录因子的碱性亮氨酸拉链,通过结合顺式作用的增强子序列调节一系列的基础和诱导表达,发挥细胞保护和抗氧化作用[18]。在静态条件下,Nrf2通过结合其抑制剂KEAP1而保留在细胞质中。如果该结合被破坏,Nrf2会转移至细胞核,并启动抗氧化基因的转录[19]。本研究发现,与对照组比较,胃炎株组和胃癌株组MGs黏膜Nrf2蛋白的表达增加,Nqo1和Ho-1蛋白的表达降低;与胃炎株组比较,胃癌株组Nrf2蛋白表达增加,Nqo1和Ho-1蛋白表达降低,差异均有统计学意义(P<0.05)。这表明,不同疾病来源的H.pylori导致胃黏膜损伤差异可能与Nrf2/Ho-1通路相关。
综上所述,不同疾病来源的H.pylori对MGs胃黏膜的损伤有明显不同,胃癌来源的H.pylori对MGs胃黏膜的损伤能力更强,这种损伤差异可能与Nrf2/Ho-1通路的激活有关。胃癌分离H.pylori菌株致病能力的具体分子机制尚有待进一步研究。
猜你喜欢
健康护理(2022年5期)2022-05-26
学苑创造·A版(2022年5期)2022-05-19
汉字汉语研究(2019年2期)2019-08-27
小学生优秀作文(低年级)(2019年5期)2019-04-25
流行色(2018年11期)2018-03-23
中国当代医药(2015年7期)2015-03-01
中医研究(2014年11期)2014-03-11
中国中医药现代远程教育(2014年20期)2014-03-01
中国中医药现代远程教育(2014年16期)2014-03-01
中国中医药现代远程教育(2014年14期)2014-03-01
