
二陈合剂质量标准提高△
2019-08-02 01:49罗镭邵喜英
中国现代中药 2019年7期
罗镭,邵喜英
1.浙江省食品药品检验研究院,浙江 杭州 310052;2.浙江省肿瘤医院,浙江 杭州 310022
二陈合剂现行标准收载于《卫生部药品标准》中药成方制剂第七册(标准编号:WS3-B-1272-93),处方由陈皮、半夏(姜制)、茯苓、甘草(蜜炙)、生姜五味药材组成,具有燥湿化痰、理气和胃之功效,临床上用于治疗咳嗽痰多、胸脘涨闷、恶心呕吐等[1]。二陈合剂现行标准较简单,仅收载性状、装量、相对密度3个检测项目,无薄层鉴别及含量测定关键质控项目。检索二陈合剂相关文献,仅检索到3篇。魏淑梅等[2]研究制定了二陈合剂的质量标准,文中建立了陈皮、半夏的薄层色谱方法。刘菁等[3]建立了二陈合剂中橙皮苷的高效液相含量测定方法。魏清[4]建立了HPLC同时测定二陈合剂中8种成分的方法。二陈合剂为2017年度国家药品标准提高品种,在相关文献[5]基础上,通过深入研究二陈合剂质量标准,综合考虑制定标准的简便性及可重复性,作者建立了二陈合剂中陈皮、甘草的薄层色谱鉴别方法,建立了二陈合剂中橙皮苷的HPLC含量测定方法,橙皮苷的HPLC含量测定方法与刘菁报道的方法有较大不同,在提取溶剂、提取方法等方面进行了优化比较。
1 材料
1.1 仪器
岛津LC-20AD型高效液相色谱仪;XPE205分析天平(瑞士梅特勒-托利多公司);Elmasonic P型超声仪(德国Elma公司);Milli-Q型纯水仪(美国Millipore公司)。
1.2 试药
二陈合剂(批号分别为:170601、160102、160103、160104、150701、150801、150802)由浙江惠松制药有限公司提供;二陈合剂(批号分别为:18070001、18070002、18070003)由太极集团重庆桐君阁药厂有限公司提供;橙皮苷(批号:110721-201617,纯度96.1%)、甘草苷(批号:111610-201607,纯度93.1%)均购自中国食品药品检定研究院;液相用甲醇为Merck公司色谱纯;其他试剂均为分析纯。
2 方法与结果
2.1 薄层鉴别
2.1.1 陈皮 用量筒量取二陈合剂20 mL置分液漏斗中,加入乙酸乙酯20 mL,充分振摇提取,高速离心使分层,吸取乙酸乙酯层置蒸发皿中,水浴低温蒸干,残渣加甲醇5 mL使溶解,滤过,滤液作为供试品溶液。另按处方配比制备除陈皮的二陈合剂阴性样品,按上述方法制备二陈合剂阴性样品溶液。再取橙皮苷对照品,加甲醇制成饱和溶液,作为对照品溶液。分别吸取上面3种样品溶液各5 μL,点于同一块用0.5%氢氧化钠溶液制备的硅胶G薄层板上,以三氯甲烷-乙酸乙酯-甲醇-水(15∶40∶22∶10)4 ℃冰箱放置过夜的下层溶液作为展开剂,展开至约8 cm,取出,晾干,喷以1%三氯化铝乙醇溶液,置紫外光(365 nm)下拍照,见图1。二陈合剂样品溶液色谱中,在与对照品色谱相应的位置上,显示一个相同颜色的荧光斑点,阴性样品溶液未显示该荧光斑点,说明缺味无干扰,方法专属性强。

注:1.橙皮苷对照品;2.阴性对照;3~9.样品图1 二陈合剂中陈皮TLC图
2.1.2 甘草 取薄层鉴别陈皮项下供试品溶液作为甘草鉴别用供试品溶液。另按处方配比制备除去甘草的阴性样品,按上述方法制备缺甘草的二陈合剂阴性样品溶液。再取甘草苷对照品,加甲醇制成每1 mL含1 mg的溶液,作为对照品溶液。分别吸取上述3种溶液各3 μL,点于同一块用1%氢氧化钠溶液制备的硅胶G薄层板上,用乙酸乙酯-甲酸-冰醋酸-水(15∶1∶1∶2)混合溶液作为展开剂,展开至8 cm,取出,置通风柜中吹干,再喷以10%硫酸乙醇溶液,在105 ℃加热至斑点显色清晰,放于紫外光灯(365 nm)下拍照,见图2。二陈合剂样品溶液色谱中,在与对照品色谱相应的位置上,显示一个相同颜色的荧光斑点,阴性样品溶液未显示该荧光斑点,说明缺味无干扰,方法专属性强。

注:1.甘草苷对照品;2.阴性对照;3~9.样品。图2 二陈合剂中甘草TLC图
2.2 橙皮苷含量测定
2.2.1 色谱条件 Agilent ZORBAX SB-C18色谱柱(250 mm×4.6 mm,5 μm);流速为1.0 mL·min-1;流动相:甲醇-4%醋酸溶液(38∶62);检测波长为283 nm;柱温为30 ℃;进样量为10 μL。
2.2.2 溶液的制备
2.2.2.1 对照品溶液制备 精密称量橙皮苷对照品14.45 mg,置100 mL容量瓶中,用甲醇溶解并定容,摇匀,即得0.138 9 mg·mL-1对照品储备液。精密吸取对照品储备液5 mL,置于10 mL量瓶中,用70%甲醇稀释至刻度,摇匀,得到0.069 43 mg·mL-1对照品溶液。
2.2.2.2 供试品溶液制备方法比较 1)精密吸取二陈合剂2 mL,置20 mL量瓶中,加入甲醇13 mL,超声处理(功率:250 W,频率:40 kHz)30 min,放冷,用甲醇稀释至刻度,摇匀,滤过,取续滤液,即得;2)精密吸取二陈合剂2 mL,置20 mL量瓶中,加入70%甲醇13 mL,超声处理(功率:250 W,频率:40 kHz)30 min,放冷,用70%甲醇稀释至刻度,摇匀,滤过,取续滤液,即得;3)精密吸取二陈合剂2 mL,置20 mL量瓶中,加入50%甲醇13 mL,超声处理(功率:250 W,频率:40 kHz)30 min,放冷,用50%甲醇稀释至刻度,摇匀,滤过,取续滤液,即得;4)精密吸取二陈合剂2 mL,置20 mL量瓶中,加入30%甲醇13 mL,超声处理(功率:250 W,频率:40 kHz)30 min,放冷,用30%甲醇稀释至刻度,摇匀,滤过,取续滤液,即得;5)精密吸取二陈合剂2 mL,置20 mL量瓶中,加70%甲醇稀释至刻度,摇匀,滤过,取续滤液,即得。
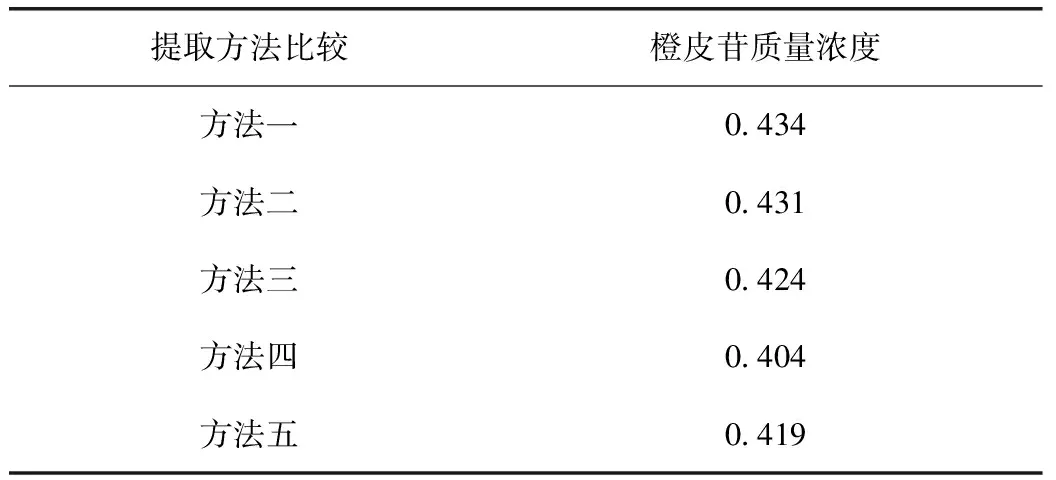
表1 供试品溶液不同制备方法测定结果 mg·mL-1
通过比较发现,甲醇和70%甲醇提取效率相当,50%甲醇、30%甲醇随着甲醇浓度降低,测定结果逐渐降低。甲醇与70%甲醇相比,加入甲醇后样品溶液有白色絮状物析出,卷成一团,样品分散不均匀,70%甲醇未发现该现象,所以最终选择70%甲醇作为提取溶剂。然后比较了是否需要超声,通过比较发现,未超声的样品含量明显偏低,最终选择提取方法为超声提取。最后确立的供试品溶液制备方法为方法二。
2.2.2.3 制备阴性样品溶液 按处方配比(除去橙皮)模拟制剂工艺,制备出缺少陈皮的阴性样品,再按2.2.2.2项下方法二,制备阴性样品溶液。
2.2.3 专属性考察 采用自动进样器依次吸取对照品溶液、供试品溶液及阴性样品溶液各10 μL,注入高效液相色谱仪系统,按2.2.1项下色谱条件进行测定,结果见图3,供试品溶液呈现与对照品溶液一致的橙皮苷色谱峰,阴性样品溶液未出现橙皮苷的色谱峰,表明阴性对照无干扰,方法专属性良好。经过与苯甲酸钠对照品比对,发现橙皮苷附近的大峰为苯甲酸钠防腐剂色谱峰。

注:A.橙皮苷对照品;B.二陈合剂样品;C.缺陈皮阴性样品;D.苯甲酸钠对照品;1.橙皮苷;2.苯甲酸钠。图3 橙皮苷对照品、苯甲酸钠对照品及二陈合剂样品高效液相色谱图
2.2.4 线性关系考察 分别精密吸取2.2.2.1项下0.069 43 mg·mL-1对照品溶液1、3、5、10 μL及0.138 9 mg·mL-1对照品储备液10 μL,按2.2.1项下色谱条件进样测定,以峰面积作为纵坐标(Y),以对照品进样量作为横坐标(X),制备标准曲线,得到线性回归方程为:Y=1 660.8X+13.089(r=0.999 9),结果橙皮苷进样量在0.069 43~1.389 μg与其对应的峰面积呈良好的线性关系。
2.2.5 精密度试验 精密吸取2.2.2.1项下0.069 43 mg·mL-1的橙皮苷对照品溶液10 μL,连续进样6针,计算出橙皮苷峰面积的RSD为0.14%,表明仪器进样误差小、仪器精密度良好。
2.2.6 重复性试验 取同一批号(批号:170601)二陈合剂样品6份,每份精密量取2 mL,置20 mL容量瓶中,加70%甲醇13 mL,超声处理(功率:250 W,频率:40 kHz)30 min,放冷,加70%甲醇稀释至刻度,摇匀,离心,取上清液,即得重复性测定用供试品溶液。按2.2.1项下色谱条件进样测定,结果二陈合剂中橙皮苷平均质量浓度为0.457 mg·mL-1,RSD为0.50%,表明供试品溶液制备方法重复性良好。
2.2.7 加样回收率试验 采用加样回收试验,称取已做重复性试验的同批号二陈合剂样品(批号:170601)6份,每份精密量取1 mL,置20 mL容量瓶中,每份分别加入橙皮苷对照品储备液(0.138 9 mg·mL-1)3 mL,再加70%甲醇11 mL,超声处理(功率:250 W,频率:40 kHz)30 min,放冷,加70%甲醇稀释至刻度,摇匀,离心,取上清液,即得回收率测定用供试品溶液。按2.2.1项下色谱条件进样测定,计算回收率,结果二陈合剂中橙皮苷的平均回收率为101.10%,6份样品的RSD为0.73%,表明方法回收率好,测定结果准确,结果见表2。

表2 加样回收试验结果(n=6)
2.2.8 稳定性试验 精密吸取同一份二陈合剂供试品溶液,分别于0、4、11、19、27、32、40 h进样,测定橙皮苷峰面积,计算各时间点橙皮苷峰面积的RSD为0.20%,表明二陈合剂供试品溶液中橙皮苷成分在40 h内稳定性良好。
2.2.9 样品测定 对收集到的10批样品,按上述色谱条件和方法测定二陈合剂中橙皮苷的含量,测定结果见表3。

表3 10批二陈合剂含量测定结果 mg·mL-1
3 讨论
3.1 甘草薄层鉴别指标的选择
《中华人民共和国药典》中甘草的薄层鉴别方法是以甘草酸铵对照品及甘草对照药材作为检测指标,笔者通过实验发现,甘草酸铵显色不清晰,二陈合剂中其他成分对其干扰严重,所以未选择甘草酸铵作为对照品。在选择甘草对照药材作为检测指标时,其有4个亮黄色的黄酮斑点,但二陈合剂样品中有个黄酮斑点与橙皮苷斑点刚好重合,缺味总是存在干扰。后改为以甘草苷对照品作为检测指标,斑点对应良好,缺味无干扰,较好地解决了甘草的薄层鉴别难题。
3.2 橙皮苷含量测定结果讨论及含量限度标准的建议
二陈合剂全国共有2家生产企业,本次实验收集到浙江惠松制药有限公司提供的7批样品及太极集团重庆桐君阁药厂有限公司提供的3批样品,根据10批样品测定结果分析,太极集团重庆桐君阁药厂有限公司样品含量较低,批间差异小;浙江惠松制药有限公司样品含量相对较高,但批间差异较大。推测可能原因是与处方工艺有较大关系,处方中陈皮提取工艺为“水煎煮3次,合并煎液,滤过”,橙皮苷在水溶液中溶解性较差,所以制剂中橙皮苷转移率较低,橙皮苷较低的转移率可能导致不同批次样品之间测定结果差异大。根据10批样品测定结果,建议暂拟订限度为:本品每1 mL含陈皮以橙皮苷(C28H34O15)计,不得少于0.30 mg。由于本次实验收集样品批次较少,待以后测定更多批次数据,再制定合理的限度。
3.3 方中其他药味薄层鉴别方法摸索
实验过程中对方中其他药味[半夏(姜制)、茯苓、生姜]的薄层鉴别方法进行过考察,半夏(姜制)、茯苓斑点较少且显色不明显,未能找到该两味药材的特征性斑点;生姜斑点较多,但是缺味始终存在干扰,故也未能建立生姜的专属性鉴别方法。
3.4 橙皮苷对照品配置问题
实验过程中发现,精密称取橙皮苷对照品15 mg置100 mL量瓶中,加70%甲醇超声溶解并定容至刻度,储备液放置过程中,会析出橙皮苷固体结晶,影响后续储备液的使用。通过摸索发现,储备液用纯甲醇配置,放置过程中不会析出橙皮苷结晶。
猜你喜欢
医学概论(2022年3期)2022-04-24
果树资源学报(2021年4期)2021-12-03
健康之家(2021年19期)2021-05-23
中国药学药品知识仓库(2021年18期)2021-02-28
国际放射医学核医学杂志(2021年10期)2021-02-28
幸福·健康版(2018年3期)2018-03-23
食品与健康(2017年9期)2017-09-13
恋爱婚姻家庭·养生版(2017年2期)2017-02-15
海峡科技与产业(2016年3期)2016-05-17
饮食科学(2015年2期)2015-09-24
