
国内家族性发作性运动诱发性运动障碍临床分析及一家系报道
2019-07-29 02:31:06刘红英李又福李现亮高庆春
广州医药 2019年4期
刘红英 李又福 张 莹 李现亮 高庆春
广州医科大学附属第二医院神经内科(广州510260)
发作性运动障碍(paroxysmal dyskinesias, PD)是一组以反复发作的、短暂性运动障碍为特征的疾病,可表现为肌张力障碍、舞蹈样动作、投掷症或手足徐动等。根据诱发因素和发病持续时间可分为4类:①发作性运动诱发性运动障碍(PKD):突然由静止到运动或改变运动形式诱发;②发作性非运动诱发性运动障碍(PNKD):自发发生,饮用咖啡、茶和酒精、疲劳等是常见诱因;③发作性过度运动导致的运动障碍(PED):在长时间运动后发作;④发作性睡眠诱发性运动障碍(PHD):在睡眠中发作。其中PKD是最常见的一种,可为遗传或散发,遗传方式大多为常染色体显性遗传,有外显不全现象[1]。
运用CNKI及万方数据库查阅文献及博士、硕士研究论文、会议论文(2004年-2018年),剔除重复者,国内共报道了家族性PKD 47个家系[2-16],结合本课题组发现的一个单纯型PKD家系共48个家系,本论文初步总结分析了国内家族型PKD的临床表现、遗传方式、辅助检查结果及治疗预后。现将我们诊治一个PKD家系的5例患者临床资料及国内家族性PKD临床特征做如下分析。
1 临床资料
1.1 临床特点(见表1)
该家系来自广东广州,表兄妹三人同时就诊,具有相似的临床表现,主诉:发作性四肢僵硬、运动不能10+年。6~7岁时起病,以突然运动,如体育课上、课堂被老师提问、过马路时等诱发,时有下肢不适感、头晕的发作先兆,发作时表现为四肢僵硬,运动不能,持续十秒钟左右能自行缓解,发作过程中意识清楚。若突然运动时患者转移注意力,有时能避免发作。每天发作次数不定,多者数十次,每次发作症状相同。发作间期正常。既往无高热惊厥病史,生长发育正常。其父辈们曾有类似症状发生,现已无发作。查体:意识清楚,智力正常,神经系统查体未发现阳性体征。辅助检查:EEG、颅脑MRI未见异常。
1.2 家系分析
该家系共11人,5人发病,男3人,女2人,起病年龄7~9岁,分析遗传方式符合常染色体显性遗传,伴不完全外显。第Ⅱ代患者未接收治疗,26~27岁时自行停止发作,病程约20年。此家系现未见遗传早现现象。遗传系谱图见图1。
1.3 诊断
单纯型家族性发作性运动诱发性运动障碍。
1.4 治疗
Ⅲ2初始服用卡马西平50 mg qd,能明显减少发作次数,但仍有发作;后加量卡马西平50 mg bid,随访2年间有少量发作,对日常生活无影响。Ⅲ1、Ⅲ3因恐惧药物副作用,未接受治疗,现每天仍有发作。
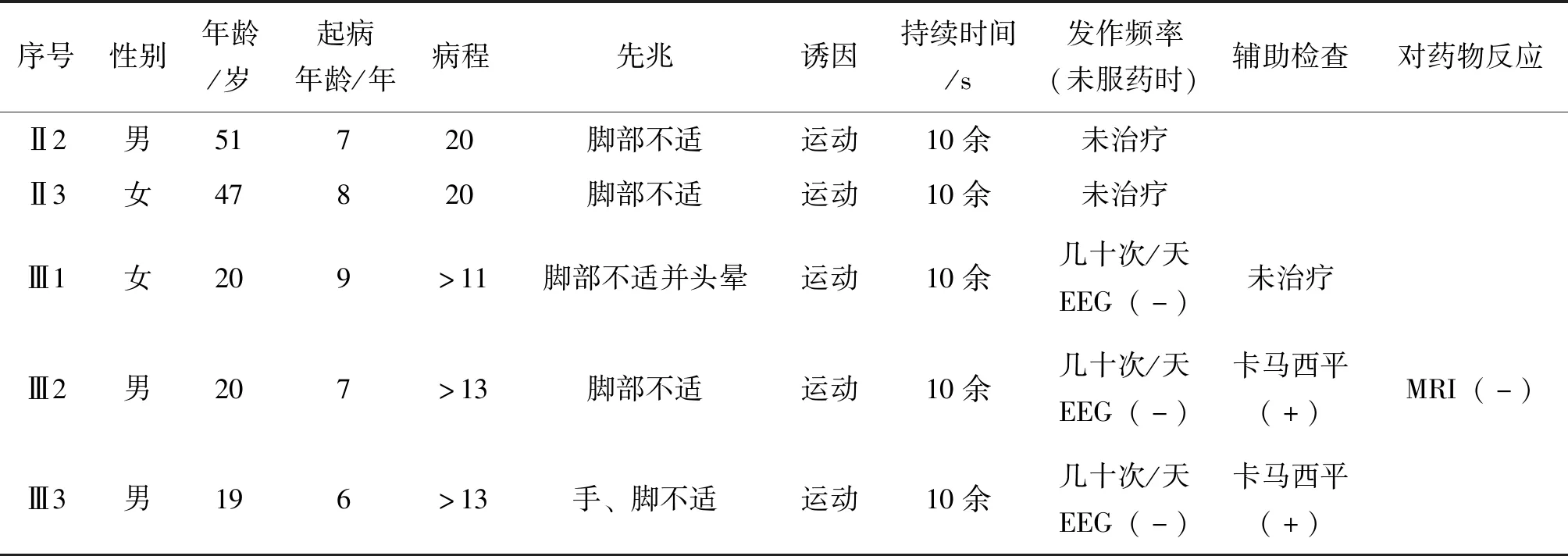
表1 发作性运动诱发性运动障碍临床特点

图1 家族性发作性运动诱发性运动障碍家系遗传系谱图
2 国内48个PKD家系分析
PKD为临床较少见的肌张力障碍类型,临床表现形式具有多样化,容易漏诊、误诊。查阅文献,在国内期刊论文、研究生论文及会议论文中报道的家族性PKD,共48个家系,191例患者,其中男130例,女61例,男:女=2.1:1。起病年龄最小1岁,最大者30岁,平均年龄10.5岁。国内报道的48个家系中,单纯型为32个家系,临床表现为发作由运动或体位改变而诱发,表现有舞蹈样动作、肢体僵硬、痉挛样动作、手足徐动及躯体扭转等,多累及一侧肢体,部分双侧肢体同时累及,发作持续时间数秒至数十秒,一般不超过1min。多数有发作先兆,转移注意力、缓慢起始运动能避免发作,发作时意识清楚,发作间期正常。复杂型家族性PKD有16个家系,其中有13个家系合并有癫痫发作,1个家系合并特发性震颤,1个家系合并Friedrich共济失调,1个家系合并偏头痛。复杂型PKD伴癫痫者,癫痫发作类型以热性惊厥、强直-阵挛发作多见。合并特发性震颤者,除了PKD的临床表现同时伴有双手震颤,渐进性加重,影响到日常生活。合并Friedreich共济失调患者4岁时出现行走不稳、行动笨拙、易跌倒,病情逐渐加重。合并偏头痛发作的PKD家系中,4例患者均合并偏头痛。体格检查:单纯型PKD患者生长发育正常,智力正常,神经系统查体无阳性体征。合并Friedreich共济失调家系先征者查体可见宽基底步态,弓形足,双眼左视时可见水平粗大眼震,四肢肌张力低,腱反射减弱,双下肢音叉震动觉消失,左侧Babinski征阳性,Romberg征睁、闭眼时均阳性,跟-膝-胫试验不准。合并特发性震颤家系先证者查体双上肢可见明显动作性震颤,频率5 Hz左右,指鼻试验欠准。辅助检查:单纯型PKD家系中行EEG或24 h视频脑电图或动态脑电图检查的患者中绝大部分是正常,部分患者发作间期可见阵发性慢活动及散在尖波、尖慢波,但发作时未见同步痫样放电。复杂型PKD家系伴癫痫者,脑电图检查可见尖波、尖慢波及棘慢波发放。头颅CT或MRI、MRA检查,有1例示透明隔囊肿,其余无阳性发现。治疗及预后:给予卡马西平或奥卡西平或苯妥英钠治疗的患者,除伴有癫痫发作之外(部分加用丙戊酸钠或托吡酯),其余全部有效,合并特发性震颤者,加用朴米酮 0.25 g bid治疗,震颤明显减轻。药物控制运动障碍发作后,部分患者停药后未再复发,若复发,再用药依然有效。
PKD随年龄增长,发作次数逐渐减少,大部分到40岁时发作次数明显减少,停止发作的年龄以家系及个体而不同,部分30岁左右时已停止发作。有部分家系显示男性患者的临床症状比女性重,自然缓解的年龄晚于女性患者。遗传特点分析:48个家系中42个家系呈常染色体显性遗传,部分伴有不完全外显现象,其余6个家系仅有一代人发病,不排除隐性遗传的可能,尚需进一步随访观察。呈显性遗传的42个家系中有10个家系有遗传早现现象,其中有9个家系为单纯型,表现为发病年龄提前或严重程度逐渐增加,1个家系为复杂型伴有婴儿惊厥,表现为以发病年龄提前为主[16],国外家系中未发现此现象。国内家族性PKD与散发性患者相比,具有发病年龄低、发病持续时间短的特点,其他临床表现无差异。
3 讨 论
虽然PKD是一种少见病,但国内自1984年陈汉白首次报道后查阅文献截止目前已报道的散发PKD约有600例,家族性PKD有48个家系,提示本病并不罕见,且此病对抗癫痫药物敏感,预后良好,因此临床医生应提高对本病的认识。2004年Bruno[17]原发性PKD新的诊断标准:①突然运动诱发的运动障碍;②无家族史者发病年龄在1~20岁间(有家族史者年龄可适当放宽);③持续时间短暂(多<1min);④发作时无意识丧失或疼痛感;⑤抗癫痫药物治疗有效;⑥排除其他器质性病变。鉴别诊断:PKD大多为原发,EEG及影像学检查无特殊表现,主要依靠临床特征来诊断,需要排除脑外伤、缺血缺氧性脑病、多发性硬化等引起的继发性PKD,首选头颅MRI及EEG。需与癫痫、肝豆状核变性、癔病、阵发性失张力性舞蹈样手足徐动症及其他类型发作性运动障碍相鉴别。
在发作性运动障碍中,绝大多数PKD患者对抗癫痫药物治疗敏感,尤其是卡马西平或苯妥英钠效果显著,若患者服用卡马西平后出现白细胞降低或皮疹过敏等明显副作用时,可试用托吡酯、小剂量奥卡西平亦有效。国外资料显示氯硝西泮、丙戊酸钠和安坦类药物无效,国内资料显示部分患者应用丙戊酸钠有效,推测可能为PKD合并癫痫患者,在临床中视患者病情酌情选用丙戊酸类药物。
2011年,国内多个研究小组率先证实了PRRT2(proline-rich transmembrane protein 2)基因为家族性发作性运动诱发性运动障碍的致病基因[18-20]。PRRT2基因定位于染色体16 P11.2,包含4个外显子,产物为富脯氨酸跨膜蛋白2,该蛋白包含2个跨膜区,分别在第269~289和318~338氨基酸之间。PRRT2蛋白是一种富含脯胺酸的跨膜蛋白,属于突触前膜蛋白,在细胞胞吐作用和神经递质的释放过程中发挥着重要的作用。PRRT2基因在尾状核、丘脑底核、苍白球、小脑等椎体外系神经组织中高表达,而其他神经组织中也有表达。迄今已在PKD患者中确定了20多种PRRT2基因突变类型,且大部分是出现在该基因的第2、3号外显子编码区的杂合型突变,包括截断突变(c.514_517 delTCTG,p.Ser172 Argfs*3;c.649 dupC,p.Arg217 Profs*8;c.972 delA,p. Val325 Serfs*12),错 义 突 变(c.796 C>T,p. R266 W;c.913 G>A,p.G305 R)等[18-20]。目前在PKD家系中发现最常见的突变是c.649 dupC (p.R217 PfsX8),且证实这个突变在8个已发表的有关PKD疾病的研究中均存在[18,20-22],c.649 dupC (p.R217 PfsX8)为移码突变,其可造成终止密码子提前出现,引起肽链截短,因此推测其导致PRRT2编码蛋白功能缺失,进而引起疾病[23]。推测在PKD临床表现中的肌张力障碍或舞蹈样动作可能与兴奋性神经递质释放增多而抑制性神经递质释放减少,这一过程可以解释钠离子通道阻滞剂卡马西平可以有效的控制临床发作。值得注意的是PRRT2基因突变包括了一个很大疾病表型谱:包括良性家族性婴儿惊厥、婴儿惊厥伴发作性手足舞蹈徐动症、偏头痛以及发作性共济失调,国内报道的复杂性PKD家系伴有婴儿惊厥、偏头痛及共济失调,推测其可能为等位基因性疾病或者是基因的多效性表达。PRRT2基因为家族性PKD患者最重要的致病基因,但不同的患者临床表现差异较大,表明PKD患者具有较大的临床表现异质性及PRRT2基因的多效性。
在目前已报道的PKD家系中,PRRT2基因突变者占91%,而在散发病例中仅为35%,提示散发性PKD的致病基因可能存在于EKD3位点。国内一研究发现PKD家系中携带PRRT2基因突变的患者较未携带PRRT2基因突变发病年龄更早、发作更为频繁,但携带PRRT2基因突变患者对卡马西平治疗全部有效,未携带PRRT2基因突变的患者仅部分有效(44.4%)[7]。因此,对于有明确家族史的患者,应首先进行PRRT2基因筛查,在筛查策略上可优先筛查热点突变c.649 dupC (p.R217 PfsX8)。
本文初步总结分析了国内48个家族性PKD家系的临床特征、遗传学特点、辅助检查、治疗预后及基因筛查等方面的资料,并报道一个单纯型PKD家系,期望对探讨家族性PKD发病机制有一定的作用。PKD在临床上属于少见病,易误诊,有资料显示39例发作性运动障碍患者,初诊正确者仅3例,鉴于PKD对药物反应性非常好,低剂量抗癫痫药物即可迅速控制发作,停药复发后再给药仍然有效,疾病预后较好,以此希望提高临床医生对该病的认识,避免漏诊、误诊。
猜你喜欢
中国神经精神疾病杂志(2022年3期)2022-07-14 02:23:42
世界科学技术-中医药现代化(2021年8期)2021-12-21 07:04:52
黄河之声(2017年6期)2017-07-01 22:35:49
中国医药科学(2015年24期)2016-03-07 15:32:46
黄河之声(2016年24期)2016-02-03 09:01:52
广东海洋大学学报(2015年4期)2016-01-13 08:39:30
听力学及言语疾病杂志(2015年5期)2015-12-24 01:47:04
首都医科大学学报(2015年4期)2015-12-16 13:00:08
小主人报(2015年5期)2015-02-28 20:43:14
郑州大学学报(医学版)(2015年2期)2015-02-27 14:50:44
