
枣遗传图谱构建研究进展
2019-07-25 12:11王中堂周广芳李新岗
核农学报 2019年7期
王中堂 张 钟 周广芳 李新岗,* 张 琼
(1 山东省果树研究所,山东 泰安 271000;2 西北农林科技大学林学院,陕西 杨凌 712100)
枣(ZiziphusjujubaMill.,2n=2x=24)为鼠李科枣属植物,原产于中国,据史料记载,在我国枣已有 7 000 余年的栽培历史[1]。枣果实具有较高的营养价值和药用价值,可鲜食、可制干,深受消费者青睐。据统计,目前我国枣栽培面积约200万hm2,2016年折合干枣年产约624.9万t,干枣产量占总产量的80%[2-3]。此外,枣果实中维生素C含量平均约为250 mg·g-1[4],环磷酸腺苷(cyclic adenosine monophosphate,cAMP)超过350 μg·g-1,高于其他同类高等植物[5-6]。枣种质资源丰富,约有840个品种,但多数品种是通过农家选优和自然实生选育而来,常规育种方法耗时费力,且目标性较弱,因此,遗传理论研究和育种技术创新,是提高定向培育新品种速度的前提。随着分子生物学技术的迅猛发展,枣基因组已广泛应用于鉴定枣树品种[7-8]、亲缘关系[9]、遗传多样性的分析[10-13]、连锁图谱的构建、数量性状位点(quantitative trait loci,QTLs)定位、分子标记辅助育种等研究[14]。利用遗传图谱可以进行QTLs,克隆目的基因,从而开展分子辅助育种,因此要实现枣基因组应用于枣树遗传育种,还需加大遗传连锁图谱的研究[15]。本文对目前国内外枣遗传图谱构建的理论基础、一般程序和方法、所取得的成果等方面进行简要概述,并对存在的问题及解决措施进行探讨,以期为开展枣农艺状候选基因筛选和分子辅助育种研究提供参考。
1 遗传图谱概述
1.1 遗传图谱构建原理
遗传作图是将基因或遗传标记,以重组型配子推算出重组率并转化而来的遗传图距为标准,顺序排列成连锁群的过程[16]。通常以厘摩(centi-morgan,cM)表示标记间的距离[17]。1913年世界上第1张遗传图谱被构建完成,用于描述果蝇的遗传特性[18],在此基础上,多个物种的遗传图谱相继被构建出来[19-24]。随着图谱构建理论和技术的提高和分子标记的发展,遗传图谱的标记数量和密度不断增加,在染色体上的分布也越来越均匀[25]。早期研究主要通过开发扩增片段长度多态性(amplification fragment length polymorphism,AFLP)、随机扩增多态性(random amplified polymorphic DNA,RAPD)等分子标记构建遗传图谱,但所构建的遗传图谱质量较低,且只能通过多个群体、多次作图才能将图谱加密,达到理想的效果[15]。微卫星(simple sequence repeat,SSR)标记是一种优良的、共显性标记,广泛应用于不同的群体研究中,但其易受到标记数量限制,也存在标记密度较低的问题[26]。研究表明,下一代测序技术(next generation sequencing,NGS)开发包括单核苷酸多态性(single nucleotide polymorphism,SNP)和插入缺失(insertion-deletion,INDEL)位点较成熟,通过大规模测序,可开发大量遗传标记位点[27]。因此,SNP标记已广泛运用于构建高密度遗传图谱、相关性状的QTL定位、基因挖掘分析等方面的研究。
1.2 枣树分子遗传图谱构建方法
枣树分子遗传图谱的构建方法是先确定亲本杂交组合,这不仅为后续构建作图群体提供亲本材料,还是群体能否构建成功的关键。然后建立作图群体构,开发合适的分子标记(AFLP、RAPD、SSR、SNP),测定标记基因型,最后分析标记基因型数据,构建遗传图谱[28]。
1.2.1 选择亲本组合 枣树亲本选择要求父母本亲缘关系相对较远,母本为雄性不育,若花期一致,父本花量大则为最佳组合[29]。当前已构建图谱的亲本组合中,母本仅有冬枣[30-32]和JMS2[33],可用资源较少。目前已报道的可作为母本的有13份种质,分别为雄性不育1号(JMS1)、雄性不育3号(JMS3)、梨枣、六月枣、酸疙瘩枣[34-35]、早脆王[36],均为无花粉的雄性不育种质;大荔圆枣、圆铃枣、广东白枣、小墩墩枣、小枣4、洪赵十月红枣和小枣25均为稳定表现为自花不实或自花可实不育而异花授粉可育率高且种仁饱满种质[37]。
1.2.2 建立作图群体 枣树与其他树种一样,均是基于双假测交(double pseudo-testcross)理论,利用F1分离群体进行遗传图谱构建[38]。但枣树花小、人工授粉杂交困难,落花落果和胚败育现象十分严重[39-41],只能通过自然授粉和人工控制自然授粉的方式获得杂交种子,从而获得实生后代[29,42]。采集实生后代和父母本(包括可疑父本)新鲜叶片,提取基因组DNA[8,43]。利用父母本(包括可疑父本)进行引物多态性筛选(表1)。对筛选引物进行PCR扩增,对扩增产物进行毛细管电泳,检测SSR位点。利用GeneMarker[8]或GeneMapper 4.0[43]软件读取毛细管电泳数据,利用FlexiBinv2[44]程序矫正读取数据。利用Cervus 3.0软件[45]对子代、母本和候选父本的等位基因数量、观测杂合度、期望杂合度和多态性信息含量,进行母本已知、父本未知的模拟分析(simulation of parentage analysis)和亲子分析(parentage analysis),在可信度95%条件下找到唯一父本的杂交后代用于构建枣树遗传作图群体[8,31-33,43,46]。利用SSR分型技术可以检测母本子代的唯一父本,且在选择亲本组合时,母本是否雄性不育则不需要再考虑,只需要考虑两亲本之间的性状差异,但如果母本雄性不育,则会极大降低SSR分型的工作量。

表1 经过筛选的多态性较好引物Table 1 Selected primers with better polymorphism
1.2.3 选择适当的分子标记,测定标记基因型 枣树遗传图谱构建利用的分子标记有AFLP[16,31]、RAPD[47-49]、SSR[30,48]和SNP[31-33]。
1.2.4 常用作图软件 可采用FsLinkageMap[31]、Join MAP[50-51]、CARTHAGENE[52]、AntMAP[53]、RECORD[54]、TMAP[55]、MSTMAP[56]、MapMarker等[57]软件进行单一作图群体构建遗传图谱研究,其中FsLinkageMap充分考虑了双亲标记的多种分离类型,主要用于构建林木全同胞家系遗传图谱[21],但该软件尚未被用于枣树遗传图谱构建。MapMarker是早期遗传图谱构建的经典软件,由Lander等[57]开发,适用于F2群体、回交群体(backcross,BC)和重组自交系群体(recombinant inbred lines,RIL)等群体作图,具有可操作性强、可定位复合性状基因等特点,但其最多只能处理1 000个分子标记,且对于异交林木群体,不能整合1∶1分离和1∶3分离的标记位点[31],因此尚未用于枣树遗传图谱构建。CARTHAGENE、AntMAP、RECORD、TMAP和MSTMAP 5种软件也均未被用于枣树遗传图谱构建研究。目前已构建的枣树遗传图谱主要采用Stam[51]开发的JionMap软件操作简单,适合F2、BC、RIL等各类群体,但需购买证书,且该软件采用最小二乘法来估算相邻标记间的遗传距离,可能会影响作图的准确性。JionMap已出现4.1版本,其功能强大,可同时分析50 000个标记,可利用锚定标记判断同源连锁群[45]。采用单一作图群体构建遗传图谱会受群体大小限制,图谱质量不高,需要将2个或多个图谱合并,研究者常采用Mergemap软件[58]将单个遗传图合并成1个基于共享位点的一致图。如Yan等[59]利用二倍体玫瑰的3个作图群体,依据26个共有SSR标记,采用Mergemap软件构建了1张一致性遗传图谱,图谱质量较单群体图谱明显提升。
2 枣树遗传图谱构建研究现状
由于遗传背景复杂,许多枣树品种存在自花结实现象,无法得到重组近交系,遗传作图较困难,但枣树遗传组成高度杂合,在杂交F1时许多遗传位点会产生分离[29-31],可采用双假测交(double pseudo test cross)策略构建图谱。其原理是两个高度杂合的亲本中, 一个亲本的杂合位点与另一亲本的杂合或隐性纯合位点在F1便发生分离,若一亲本为杂合位点,另一亲本为隐性纯合位点, F1呈 1∶1 分离, 相当于测交,可用于构建亲本的图谱; 当双亲均为杂合位点时,呈 3∶1(显示标记)或 1∶2∶1(共显性标记)分离,可用于构建两个亲本共同的分子连锁图及判断双亲间的同源连锁群[38]。 目前已有利用双假测交在苹果[38]、葡萄[60]、梨[61-62]等果树上成功构建1张或多张遗传图谱,并开展大量QTL定位的研究报道,因此,枣树也可利用该方法构建图谱[63-64]。
2.1 枣树遗传图谱构建
我国枣树分子遗传图谱的研究较晚,鹿金颖[49]于2003年以临猗梨枣(父本)×冬枣(母本)72株F1为作图材料,利用AFLP标记构建了枣树的第1张遗传连锁图,且该连锁图开发了28个AFLP标记,分布于7个连锁群上,覆盖基因组总长度为458.66 cM,平均图距为16.38 cM,为枣树遗传图谱构建研究提供了模板,但进一步研究发现该图标记较少,图距较大,连锁群与枣树染色体树并不对应,不能用于后续QTL精准定位研究。为构建更高密度遗传图谱,申连英[30]扩大了作图群体,利用161株临猗梨枣(父本)×冬枣(母本)F1为作图群体,构建了1张密度更高的遗传连锁图谱,该图谱包含333个多态性位点,分布于14个连锁群上,覆盖基因组总长度为1 237.4 cM,标记间平均图距3.9 cM,其中大于 10 cM的标记间隔有21个。虽然图谱多态性位点和覆盖基因组总长度均增加,且标记间平均图距减小,可进行QTL定位研究,但同时其大于10 cM的标记间隔较多,定位准确度较低,且遗传图谱连锁群与枣树染色体树也不对应,图谱质量有待提高。齐靖等[47-48]采用RAPD和SSR标记,以150株临猗梨枣(父本)×冬枣(母本)F1为作图群体,整合申连英[30]开发的419个AFLP标记,共同进行连锁分析,构建了1张包含35个RAPD标记和388个AFLP标记,15个连锁群的遗传连锁图谱,该图覆盖枣基因组长度为1 309.4 cM,标记间平均距离为3.1 cM,大于10 cM的标记间隔仅14个。与申连英[30]构建的图谱相比,覆盖基因组总长度增加72 cM,标记间平均图距缩短0.8 cM,大于10 cM的标记间隔减少7个,图谱饱和度显著提高,但图谱连锁群数目与枣树染色体仍不对应,其原因可能是以上3张图均使用传统的AFLP、RAPD等标记,缺乏基因组测序的相关信息,图谱上可用标记数量较少、标记分布密度较低、连锁群上多具有较大孔隙,难以进行全面的性状QTL研究[31]。因此,开发新的SSR标记用于枣树谱图构建很有必要。许莉斯[65]以150株临猗梨枣(父本)×冬枣(母本)F1为作图群体,开发了4个SSR标记,整合申连英[30]对此群体研究得出的333个AFLP标记,进行共同连锁分析,构建了包含336个AFLP标记和4个SSR标记遗传图谱,该图谱包含14个连锁群,4个SSR标记分别位于SLG3、SLG6、SLG9和SLGl3连锁群,但与齐靖等[47-48]构建的图谱相比减少了52个AFLP标记,且该图谱连锁群与枣树染色体的数目仍不对应,其原因可能是染色体交换频繁,标记间的连锁关系不稳定或者标记数量有限,标记间隙较大,存在空缺区段,导致同一条染色体上的连锁群不能连锁。
随着分子测序技术的迅猛发展,Zhao等[33]以105株邢16(父本)×雄性不育2号-JMS2(母本)F1为作图群体,利用RAD Tag测序技术开发了1张枣树高密度遗传连锁图谱,该图谱整合了2 748个SNP标记,分布在12条连锁群上,基因覆盖总长度为913.87 cM,平均标记间距离为0.34 cM,该图谱较前期构建的图谱多态性位点增加近8倍,标记间平均图距更加精细。Zhang等[32]以145株中宁圆枣(父本)×冬枣(母本)F1为作图群体,采用基因分型简化测序(genotyping by sequencing, GBS)技术,也构建了1张由12个连锁群组成的高密度遗传图谱,获得了2 540个SNP标记,图谱总长度为1 456.73 cM,平均遗传距离为0.88 cM,该图谱较Zhao等[33]构建的图谱覆盖总长度增加542.86 cM,但SNP标记数量减少208个,平均标记间距离增加0.54 cM。张振东[31]以99株映山红(父本)×冬枣(母本)F1为作图群体,采用限制性酶切位点相关的DNA测序(restriction-site associated DNA sequencing,RAD-seq)技术,也构建了1张枣树高密度遗传图谱,包含4 669个标记,其中SSR标记46个,SNP标记4 137个和InDel标记486个,12个连锁群(LG1~12),覆盖基因组总长度为2 643.79 cM,平均遗传距离为0.57 cM,该图较Zhang等[32]图谱覆盖总长度有所增加,SNP标记数量多,但平均遗传距离较Zhao等[33]图谱大。这3张图谱是截至目前基因组覆盖最全面、标记密度最大的枣树遗传图谱。已发表的枣树遗传图谱详见表2。
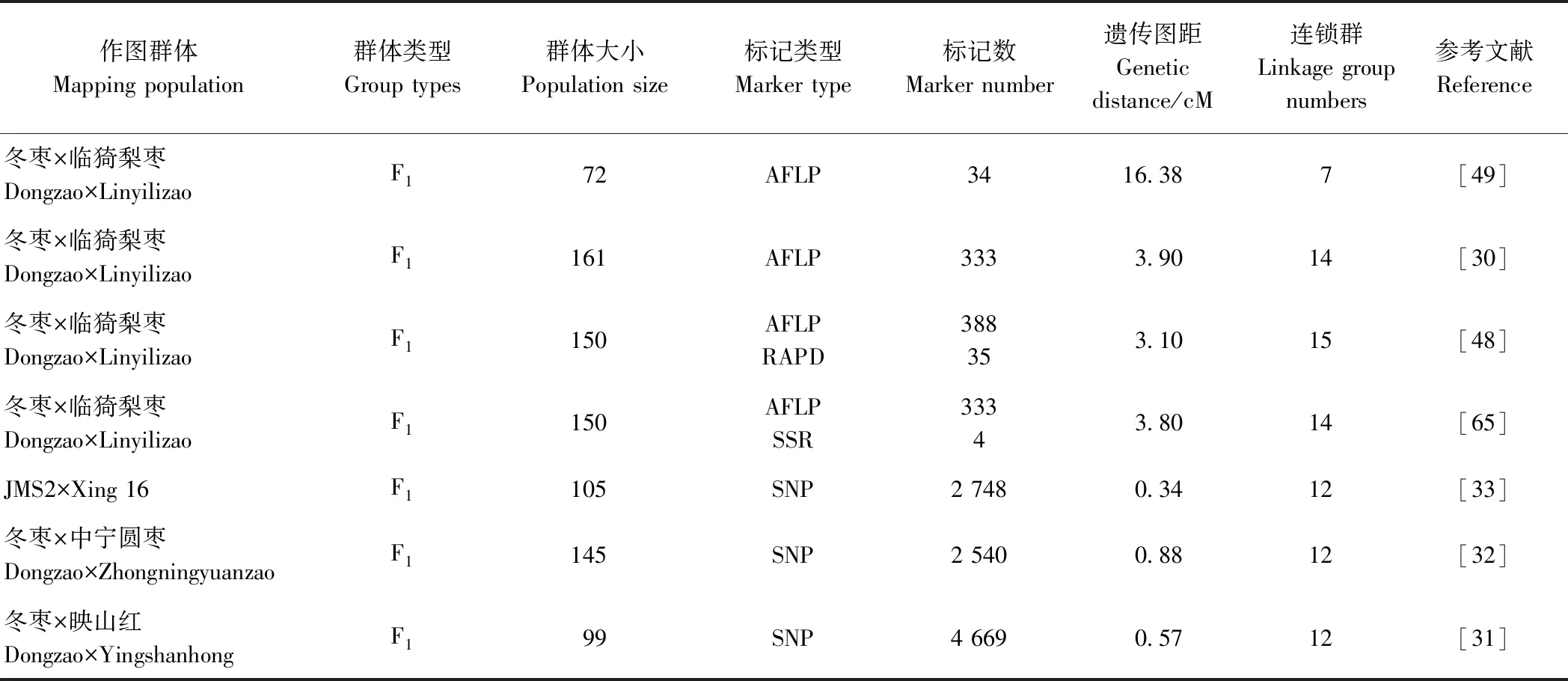
表2 已构建的枣树遗传图谱Table 2 The genetic map of jujube was construted
2.2 枣树性状QTL定位研究
构建枣树遗传连锁图谱的目的之一是进行重要性状QTL定位研究,申连英[30]、齐靖[47]利用早期开发的遗传图谱,对枣树树干、二次枝、叶片性状、叶部病害、叶片营养、枝条抗寒性等方面进行了初步的QTL定位研究(表3),在生长相关性状方面,检测到3个QTLs与2年生主干节间长度相关,7个QTLs与2年生二次枝节间长度相关,6个QTLs与3年生二次枝节间长度相关,6个QTLs与2年生枣吊节间长相关,8个QTLs与3年生枣吊节间长相关。在叶片性状方面,检测到7个QTLs与叶片长度相关,9个QTLs与叶片宽度相关,1个QTLs与枣吊叶面积相关,3个QTLs与二次枝叶面积相关,2个QTLs与叶缘锯齿数相关。在叶片营养方面,检测到3个QTLs与叶片全N含量相关,7个QTLs与叶片全P含量相关,3个QTLs与叶片全K含量相关。枝条抗寒性方面,检测到7个QTLs与-25℃相对电导率变化系数相关,9个QTLs与-35℃相对电导率变化系数相关。叶部病害方面,检测到5个QTLs与叶片失绿相关,5个QTLs与枣锈病相关,11个QTLs与枣褐斑病相关。许莉斯[65]利用已构建的AFLP连锁图谱对枣果实纵径、横径、果形指数、单果重进行了QTL定位分析。孙拥军等[66]利用许莉斯[65]构建的AFLP连锁图谱,对中心干直径、二次枝着生角、二次枝弯度、二次枝曲度性状进行了QTL定位分析,但定位到的QTL数量较少,能够解释表型变异的比率较低。
注:“-”表示无。
Note:‘-’ indicates no.
张振东[31]利用整合图谱,对株高、地径、叶面积和抗寒性4个表型性状进行QTL定位,利用MapQTL 6.0的Kruskal-Wallis模型,检测到117个QTLs,将控制株高、地径和叶面积等生长相关性状的QTLs定位到3号连锁群48.18~85.23 cM区域和134.80~195.72 cM区域,与抗寒性相关的QTLs定位到第7号连锁群0~45.41 cM区域,由于该图谱群体数量只有99株,如扩大作图群体,该研究QTL定位还能更加精确。早期图谱质量较低,调查性状较少,真正与果实品质有关的精细QTL定位尚未开展,已构建的高密度遗传图谱也还未进行QTL定位研究[32-33]。
2.3 枣树遗传图谱构建中存在的问题
枣树高密度遗传图谱的构建为枣树遗传育种提供了新的工具(表2、3),目前已构建的枣树遗传图谱,基因组的覆盖度和图距质量越来越高,为实际应用提供了基础,但也存在作图群体较小的问题。遗传标记数目的多少与这些标记基因在图谱上的定位是否准确共同衡量遗传图谱的质量[67]。在图谱构建时,要想确保检测到减数分裂时的重组事件,需要一定数量的作图群体。构建框架图谱要求的理论群体数量应大于150个[68],当需要检测到的重组分数达到0.3,LOD值为3.0时,则需要不低于300个的群体[61-62]。目前已构建的枣树遗传图谱[30-33,47-48,65]使用的分离群体数量均少于300个。若遗传图谱用于QTLs定位,则要求标记的平均图距应低于10 cM,若用于基因克隆,则标记间的平均图距低于1 cM[25]。当前已构建的枣树遗传图谱,大部分可以用于QTLs定位,只有3张图谱可用于目的基因克隆。此外,已进行的QTL研究中,存在标记数量少,定位不准确的现象,原因可能是图谱自身质量不高,数量性状调查数据较少,为提高定位的准确性,应采取多年、多地点性状调查。
3 展望
目前已建成的枣树遗传连锁图谱由于各种限制,还未全面进入应用阶段。接下来可从以下几个方面进行研究,以扩大枣树遗传图谱的应用范围。
3.1 优化亲本组合
当前已构建图谱的亲本组合为冬枣(母本)×临猗梨枣(父本)、JMS2(母本)×邢16(父本)、冬枣(母本)×中宁圆枣(父母)、冬枣(母本)×映山红(父本),母本只有冬枣和JMS2。已报道可作母本的资源有13个,分别是无花粉的枣雄性不育种质:雄性不育1号(JMS1)、雄性不育3号(JMS3)、梨枣、六月枣、酸疙瘩枣[34-35]、早脆王[36];稳定表现自花不实或自花可实不育而异花授粉可育率高且种仁饱满种质:大荔圆枣、圆铃枣、小墩墩枣、小枣4、小枣25、广东白枣、洪赵十月红枣[37]。但以上品种还未用于图谱构建,因此应利用分子标记技术加大对性状明显的优良品种的自交不亲和鉴定,发现更多适用于母本的品种,以扩大品种选择范围。
3.2 扩大作图群体
增加作图群体数量是提高遗传图谱质量的重要措施之一[68]。目前已构建的枣树遗传图谱[30-33,48]群体数量均低于150株(除申连英[30]、齐靖等[47-48])。图谱要满足精细QTLs定位,分离群体数量应为200~300个[69],而当前图谱群体数量明显不足,因此利用SSR分型技术[8,43]进行子代鉴定对扩大作图群体至关重要,可以明显提高图谱质量。因大多数拥有共同母本(冬枣),利用当前已构建图谱,基于相同的参考基因组构建图谱,根据2套或多套图谱的共享位点,利用MergeMap[58-59]软件进行2个或多个图谱整合,构建一致性遗传图谱,提升图谱质量。
3.3 研发适合枣树自身特点的作图理论和方法
农作物多利用高世代家系进行作图,这些家系标记的分离类型简单,而大多数枣树为自花结实,无法采用近交物种的F2或回交子代。当前,枣树和其他果树一样,均是利用双假测交理论,以杂交F1为作图群体,而枣树花小、人工授粉杂交困难,落花落果和胚败育十分严重[39-40],获得定向杂交的F1困难,且获得的群体在生长过程中易受枣疯病侵害,可能发生丢失,因此,在获得杂交群体后应及时对各个体进行备份。此外,还应加快研发适合枣树自身特点的作图理论和方法,加速枣树遗传图谱的构建。
3.4 加快利用已构建图谱的速度
目前虽已构建了3张枣树高密度遗传图谱[31-33],但尚未有利用图谱进行果实性状QTL定位的研究报道。未来应尽快开展QTL定位研究,以期为重要性状目标基因克隆奠定基础。
3.5 加强资源共享
遗传图谱的构建需要合适的亲本组合,这就要求资源保存单位之间能够资源共享,共同利用,合作开发。目前已构建的遗传图谱利用群体的亲本组合中,母本均为冬枣[30-32,48-49](除JMS2[33])。遗传图谱构建单位可以开放资源,依据作图群体母本相同,基于同一个参考基因组构建图谱,根据每套图谱的共有位点,进行图谱整合,对现有图谱加以利用。此外,利用西北农林科技大学[3]和河北农业大学[70]的枣全基因组测序数据,可以进行标记开发,增加标记密度,并验证标记连锁关系是否正确。
猜你喜欢
农业科技通讯(2023年1期)2023-02-12
中学生数理化·八年级物理人教版(2021年11期)2021-12-06
中学生数理化·八年级物理人教版(2020年10期)2020-11-26
中学生数理化·八年级物理人教版(2017年10期)2018-01-22
现代园艺(2017年21期)2018-01-03
中国种业(2016年11期)2016-12-01
中国稻米(2016年5期)2016-10-28
读写算(中)(2016年11期)2016-02-27
茶叶通讯(2014年1期)2014-02-27
种子科技(2012年1期)2012-01-23
