
快速起效抗抑郁药研发策略:单胺(5-HT)-非单胺(Glu/GABA)长反馈神经环路候选假说的提出
2019-07-24 09:31:14李云峰
中国药理学与毒理学杂志 2019年3期
李云峰
(军事科学院军事医学研究院毒物药物研究所,抗毒药物与毒理学国家重点实验室,神经精神药理学北京市重点实验室,北京100850)
抑郁症属于21世纪重大疾病,因其具有高发 病、高自杀、高复发、高致残率和低识别、低就诊、低治疗率等特点已成为全球性严重的公共卫生问题和突出的社会问题[1]。世界卫生组织(WHO)预测到2030 年,抑郁症将成为疾病总负担排名首位[1],目前,抑郁症在我国已成为疾病总负担第二大疾病[2]。现有临床抗抑郁药以“单胺策略”药物为主,最广泛的一线用药主要包括选择性5-羟色胺(serotonin,5-hydroxytryptamine,5-HT)重摄取抑制剂(selective serotonin reuptake inhibitors,SSRI)、5-HT/去甲肾上腺素(NE)双重重摄取抑制剂(5-HT and norepinephrine reuptake inhibitors,SNRI)、NE 能和特异性5-HT 能抗抑郁药(noradrenergic and specific serotonergic antidepressants,NaSSA)和三环类抗抑郁药(tricyclic antidepressants,TCA)等。总体来说,这些药物可有效治疗抑郁症和焦虑症,但大多都存在起效时间延迟(2~6 周)、有效率不高(50%~70%)和缺乏认知改善甚至损害认知、导致性功能障碍及自杀倾向等较严重问题。因此,发展快速起效、兼可增强认知和低毒副作用新药是目前全球性重大需求。就我国而言,目前抗抑郁药市场长期被国外原研的化药所占据(约90%市场),尚无Ⅰ类创新药物上市。本文结合国内外最新进展和本实验室前期研究,管窥快速起效抗抑郁药的研发趋势和策略,并提出单胺-非单胺,即“5-HTGlu/GABA长反馈神经环路”候选假说和候选策略,供商榷和进一步深入研究。
1 抗抑郁药60年研发历程
1.1 抗抑郁药发展60年
自从1957 年第一个真正意义上的抗抑郁药丙咪嗪被偶然发现以来,至今已有60余年历史。其总体是一个新药物—新理论的循环发展的过程,大体可分为起效慢/副作用大、起效慢/副作用小和起效快/副作用小3个阶段(图1)。①起效慢/副作用大:以TCA 类和单胺氧化酶抑制剂(monoamine oxidase inhibitors,MAOI)类药物代表。绝大部分TCA药理机制是抑制5-HT/NE重摄取,提高突触间隙5-HT/NE水平,但存在心脏毒性和认知损伤等较大不良反应,可能与其抗组胺和抗胆碱作用并阻断Na+通道和L 型Ca2+通道有关。②起效慢/副作用小:以临床一线的SSRI,SNRI和NaSSA为代表,前二者机制是通过选择性抑制5-HT/NE重摄取,增加突触间隙5-HT/NE 水平,其疗效与TCA 相当,但较之于TCA 耐受性好得多,仍存在导致自杀风险、性功能障碍和认知损伤等不良反应;NaSSA 是α2受体拮抗剂和特定5-HT 受体(包括5-HT2A,5-HT2C和5-HT3)拮抗剂,通过阻断α2受体,增强脑内NE能和5-HT能神经传导,尤其是中缝核与海马5-HT1A神经传导(也被称为5-HT1A间接激动剂),其耐受性更好,起效相对较快,2周可产生最大疗效。③起效快/副作用小:目前的主流研发目标和方向之一,以近10 余年美国FDA 批准上市的仅有的2 个全新结构5-HT能多靶标新药为代表,即维拉佐酮(vilazodone,SSRI;5-HT1A部分激动剂,2011年上市)和沃替西汀(vortioxetine,SSRI;5-HT1A和5-HT1B激动剂;5-HT1D,5-HT3和5-HT7拮抗剂,2013 年上市),前者具有一周内起效和低性功能障碍等特点[3-4],后者具有低性功能障碍和潜在增强认知作用等特点[5-6]。
总体而言,基于经典的“单胺假说”,20 世纪八九十年代(具体是从1986 年氟西汀上市到2004 年度洛西汀上市)的20年掀起了抗抑郁新药研发的第一次浪潮,现有一线主流药物均在该时期被研发出来。2016年后,美国FDA批准至少6个突破性药物资格或快速通道药物,其中的S-氯胺酮(S-ketamine,S-Ket)和别孕烯醇酮(brexanolone,SAGE-547)已于2019 年上市。我们可乐观地预见,基于中枢兴奋性调控的非单胺策略,抗抑郁药研发将掀起第二次浪潮。
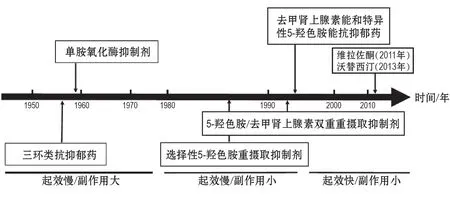
图1 临床各类主要抗抑郁药发展的简要历程.
1.2 抗抑郁药研发策略60年
第一代TCA 和MAOI 类抗抑郁药的发现直接催生了经典“单胺假说”的提出(1965年),认为抑郁症发生与脑内突触间隙NE 和5-HT 功能低下有关[7-9]。该假说指导了现有主要一线药物的研发,但无法解释抗抑郁药起效延迟、仅对部分患者有效等一系列问题。现代“单胺假说”认为,抗抑郁药作用与单胺神经元突触前膜自身受体和非单胺神经元上的异源受体(如5-HT1A/1B和α2受体)适应性调节有关,服用SSRI和SNRI等一线药物通过提高突触间隙5-HT/NE的升高,导致了单胺能自身受体和异源受体失敏,从而使5-HT/NE水平持续释放并产生抗抑郁疗效。这一失敏过程需要2~4周,这至少部分揭示了抗抑郁药起效延迟的问题[10]。因此通过优先使5-HT1A等自身和异源受体快速失敏(如5-HT1A部分激动剂),可达到加快起效并减少现有药物的不良反应的目的。近年上市的维拉佐酮支持了该假说[10-11],但该假说的主要以单胺调节层面解释快速抗抑郁的机制,忽略了快速抗抑郁的非单胺能机制(图2)。
氯胺酮(ketamine,Ket)和东莨菪碱的研究结果揭示非单胺靶标〔如GABA 能和谷氨酸(glutamate,Glu)能等调节〕在快速抗抑郁中具有更重要的调节作用[12-13]。基于神经元兴奋性调节的非单胺策略正在成为快速抗抑郁药研发的重要方向。基于该趋势,可预见未来会形成单胺-非单胺结合的多靶标全新策略,这可能是未来新药研发的必然趋势。此外,有关抑郁症发病机制较为公认的假说还包括下丘脑-垂体-肾上腺轴(hypothalamic-pituitary-adrenal axis,HPA)负反馈障碍假说、神经营养假说、脑内奖赏通路障碍假说和神经炎性损伤假说等[10]。这些假说也主要集中于非单胺层面,尽管有大量的研究,但至今尚无基于这些假说的新药上市。
抑郁症的关键靶标与神经环路关系远未阐明,抗抑郁创新药研究尚需解决很多科学问题。目前一线抗抑郁药主要基于经典“单胺假说(策略)”研发出来,但存在诸多缺陷;而约30 年来一直无基于“非单胺策略”(如HPA轴负反馈障碍和炎性损伤假说等)的新药上市,可以说抗抑郁药研发长期处于两难的困境,这也成为近年来国际药企纷纷退出抗抑郁新药研发的原因之一。直到2019年3月,首款非单胺能新机制抗抑郁药S-Ket鼻喷剂被美国FDA批准上市,用于治疗难治性抑郁症,为非单胺策略研发点燃了新希望。然而由于该药存在拟精神和致成瘾潜在不良反应受到严格管控,使其临床应用受限。我们认为,现阶段新药研发策略主要有2 个切入口:①立足优化的多靶标单胺策略(现代单胺策略),采用单胺能多靶标设计,研制快速起效、低毒副作用新药,满足临床需求;②突破现有单胺策略的框架,基于非单胺策略发现全新机制的新一代快速起效药物。这两方面代表着目前国内外抗抑郁药研发现状和阶段(图2)。
2 快速起效抗抑郁药的研发策略
2.1 优化的多靶标单胺策略

图2 抗抑郁药主要研发策略及其趋势.
优化的多靶标单胺策略(即现代单胺策略)仍是目前快速低毒抗抑郁药研发的主流方向,其主要包括5-HT 能多靶标调节药物、5-HT/NE/多巴胺(dopamine,DA)三重重摄取抑制剂、DA 受体激动剂、非典型抗精神病药与抗抑郁药联合药物等,其中以维拉佐酮和沃替西汀为代表的5-HT能多靶标调节药物因其起效相对更快和低毒副作用等特点尤其引人关注,二者都涉及SSRI 活性以及5-HT1A和5-HT1B等5-HT自身受体调节[10,14-15]。事实上,这些受体也是源于中缝核的5-HT调控前额皮质(prefrontal cortex,PFC)和海马等效应脑区的主要受体,即两药在通过SSRI提高5-HT水平的同时,也具有针对非单胺能神经元(GABA 中间神经元和Glu 锥体神经元)的直接调节机制[14-15]。由于两药都具有SSRI和5-HT1A激动活性,因此,如下以5-HT1A受体为例进行阐述多靶标单胺策略。
脑内5-HT 神经元集中于中缝背核(dorsal raphe,DR),其神经末梢长投射到PFC和海马等抑郁相关脑区,通过5-HT1A,5-HT1B,5-HT2,5-HT3,5-HT4,5-HT6和5-HT7等异源性受体调控这些脑区功能[16]。5-HT1A是脑内含量最丰富的5-HT受体,它既是突触前自身受体(位于DR 的5-HT 能神经元上,调控5-HT 释放),同时也是突触后受体(位于PFC/海马等脑区非5-HT 神经元上,介导抗抑郁行为)。PFC 是抗抑郁重要脑区之一,采用深部脑刺激(deep brain stimulation,DBS)在PFC 在大鼠上可产生最强的抗抑郁效应[17]。在PFC 中5-HT1A主要表达于Glu 能锥体神经元和GABA 中间神经元上,是调控PFC中(如第Ⅴ层)锥体神经元兴奋性的关键受体[18-19];低剂量(而非高剂量)的8-OH-DPAT增加锥体神经元的点燃效应且减弱GABA 神经元活性。相关研究提示,低剂量5-HT1A激动剂优先作用于GABA 神经元上5-HT1A,进而增强锥体神经元兴奋性[16,20]。综上所述,5-HT1A受体实际上参与了单胺和非单胺系统的双重调控。
2.2 多靶标单胺策略新药:盐酸羟哌吡酮和盐酸阿姆西汀
盐酸羟哌吡酮(YL-0919)是本实验室研发的具有原创化学结构的1.1 类抗抑郁新药(目前处于临床Ⅱ期)。该药是强效的SSRT、5-HT1A受体部分激动剂和5-HT6受体全激动剂。灌胃给药具有快速起效的抗抑郁、抗焦虑和促认知活性[21-23]。其主要药理特点表现在如下4点:①新的靶标组合且活性更强,YL-0919 选择性地与5-HT1A,5-HT6和5-HT 转运蛋白均有高亲和力结合;同时抑制5-HT 重摄取,且该作用强于氟西汀和度洛西汀;活体脑微透析发现,该药可持续提高海马5-HT 水平4 h 以上,且该作用强于氟西汀;GTPγS 实验证实其为5-HT1A部分激动剂和5-HT6全激动剂,其5-HT1A强度和最大效能均强于维拉佐酮,但维拉佐酮无5-HT6受体活性[21-23]。②抗抑郁起效更迅速,慢性应激大鼠模型灌胃给予3 d 即可产生抗抑郁作用,且7d 内快速增强海马神经可塑性,而氟西汀则要3 周[21,24]。③该药兼具有增强认知活性,而度洛西汀、氟西汀和维拉佐酮等均无此作用[23]。④该药不导致性功能障碍,而氟西汀可导致显著的性功能障碍[21]。该药化学结构与美国FDA 批准上市的维拉佐酮完全不同,且具有结构简单、合成工艺简洁和成本低廉等特点。
近期研究发现,单次给药予羟哌吡酮可激活PFC中Glu锥体神经元兴奋性,而氟西汀无此作用,并且5-HT1A受体介导了该上述作用(待发表),这与Ket 快速激活锥体神经元并导致Glu 突释是一致的。进一步采用脑片记录发现,该作用是由于该药优先作用于GABA 神经元上5-HT1A,解除了其对锥体神经元的抑制作用产生的,提示5-HT1A部分激动活性是该药快速起效的重要机制(发表中)。进而采用大鼠慢性应激实验发现,ig给予该药(2.5 mg·kg-1)3~5 d即可快速起效产生抗抑郁作用(而氟西汀要20 d),同时显著增强PFC 中脑源性神经营养因子-哺乳动物西罗莫司(雷帕霉素)靶蛋白(brain derived neurotrophic factor-mammalian target of sirolimus,BDNF-mTOR)通路及其介导的突触可塑性[24];另一方面,5-HT6激动活性介导了该药的快速的增强认知作用[23]。事实上,5-HT6分布主要限于中枢神经系统,且主要位于PFC/海马等脑区的非5-HT 神经元上。近年来,因5-HT6配体同时兼有促认知和抗抑郁作用引起药学家极大地兴趣[25],但其精细调控机制研究较少,该药为此提供了证据和工具。
基于以上研究,羟哌吡酮快速起效可能是由于直接、同时激活了5-HT 单胺系统和Glu 系统,作为SSRI、5-HT1A部分激动剂和5-HT6全激动剂,其一方面通过作用于中缝核5-HT 神经元上的5-HT1A自身受体使其快速失敏并抑制5-HT 重摄取,导致更高的5-HT 合成释放进入PFC;于此同时,还通过5-HT1A/6受体激活Glu锥体神经元,导致BDNF释放及BDNF-mTOR通路激活,快速增强PFC锥体神经元的树突复杂性和突触可塑性[26]。该研究提示,调节效应脑区的非单胺机制是快速起效的重要环节。
本课题组前期设计和发现的另一个新药是SNRI 类的盐酸阿姆西汀(071031B),该药具有起效较为快速、肝毒性低和生物利用度高的特点[27-29]。5-HT/NE 能神经投射包括海马和PFC 等抑郁症相关脑区,研究表明,同时提高5-HT 与NE神经传递有望一定程度上实现快速起效的目的。如非临床研究发现,舍曲林(属于SSRI)与瑞波西汀(属于NRI)合用,比2 药单用起效快[30]。作为第1 个上市的SNRI 的文拉法新与舍曲林相比,其疗效和起效速度都明显提高[31]。
盐酸羟哌吡酮与盐酸阿姆西汀迄今已获得国家食品药品监督管理总局1.1 类抗抑郁化药临床批件并开展Ⅰ和(或)Ⅱ期临床研究。这2个药物的成功发现,进一步证实通过“优化的单胺策略”进行合理的靶标组合,对快速起效抗抑郁药的发现具有重要借鉴意义。
3 非单胺靶标策略
基于非单胺策略发现快速起效抗抑郁新机制和新靶标是长期以来的研究热点。研究多年的磷酸二酯酶4抑制剂、NMDA受体拮抗剂,促肾上腺皮质激素释放因子1(corticotropin releasing factor 1,CRF1)受体拮抗剂和神经激肽1(neurokinin-1,NK1)受体拮抗剂、糖皮质激素激素功能抑制剂等具有显著的抗抑郁作用[10],进一步证实非单胺靶标在抑郁症治疗中的重要作用。尽管该领域已研究了约30 年,长期以来一直未见美国FDA 批准的新药上市。近年来,基于Ket 和东莨菪碱等快速起效抗抑郁药的研究,为该领域点燃了极大热情。尤其是S-Ket 鼻喷剂被批准上市用于治疗难治性抑郁症,为快速抗抑郁治疗开辟了新的时代。目前,至少有6个药物雷帕替奈(rapastinel),S-氯胺酮,AV-101,别孕烯醇酮,SAGE-217,AXS-05)已获得美国FDA突破性药物资格或快速通道资格(其中S-Ket 和别孕烯醇酮已上市),这些药物主要是NMDA 受体或GABAA受体的调节剂[32-33],这使兴奋(Glu)-抑制(GABA)平衡的调控成为重要非单胺药物研发策略,NMDA受体拮抗剂或者GABAA受体正向变构调节剂成为未来代表方向(表1)。
3.1 NMDA受体拮抗剂或部分激动剂
自2000 年Berman 等[34]首次报道NMDA 受体拮抗剂Ket 具有抗抑郁作用以来,大量临床证据表明,Ket 确可发挥快速而持久的抗抑郁作用。单次给予低剂量Ket 数小时即可产生抗抑郁疗效(持续约1 周)[35-36],且快速减少自杀愿望[37-38],尤其令人惊喜的是其对难治性抑郁症患者也有效[38]。然而,由于Ket的拟精神病及成瘾潜能使其临床应用受到限制。最近Johnson & Johnson 公司研发的经鼻给药的S-Ket 已获批上市,该药较之于R-Ket 是NMDA受体亲和力更高的阻断剂。尽管其与Ket有类似副作用,但其临床试验结果令人振奋[32,35-38]。可以说,Ket 快速、持续和高效(对难治性抑郁症有效)抗抑郁作用的发现是抑郁症领域60年来重要突破。目前对Ket 的研究主要集中在手性药物、代谢产物和结构类似物研发和快速抗抑郁作用基础研究。如Ket 代谢物2R,6R-HNK 产生快速持续抗抑郁作用,同样也无感觉运动门控等不良作用[39-40]。该类化合物的行为效应可被BDNF 功能阻断抗体所阻断,或PFC 注射mTORC1 阻断剂西罗莫司所阻断,提示其与Ket有着类似的机制[41]。然而,令人奇怪的是,其他非选择性NMDA受体拮抗剂如美金刚或拉尼西明(lanicemine)在临床试验中无效,这其中原因还不清楚,可能与剂量或NMDA阻断活性不够有关[32,42]。
Ket 具有不同于传统假说的独特机制,其可快速增加PFC 突触数量和功能且逆转抑郁症的突触病理改变[43]。事实上,慢性应激导致啮齿类动物海马和PFC等脑区神经元突触丢失及树突萎缩,脑成像研究证实抑郁患者的这2 个脑区体积缩小、突触减少,而单次给予Ket 快速增加PFC 锥体神经元突触功能和数量,且快速逆转慢性应激导致的这些神经元突触的丢失[43]。Ket 给药2 h 可导致突触蛋白(包括GluA1)水平增加,这与Ket 抗抑郁起效时间一致的[44]。
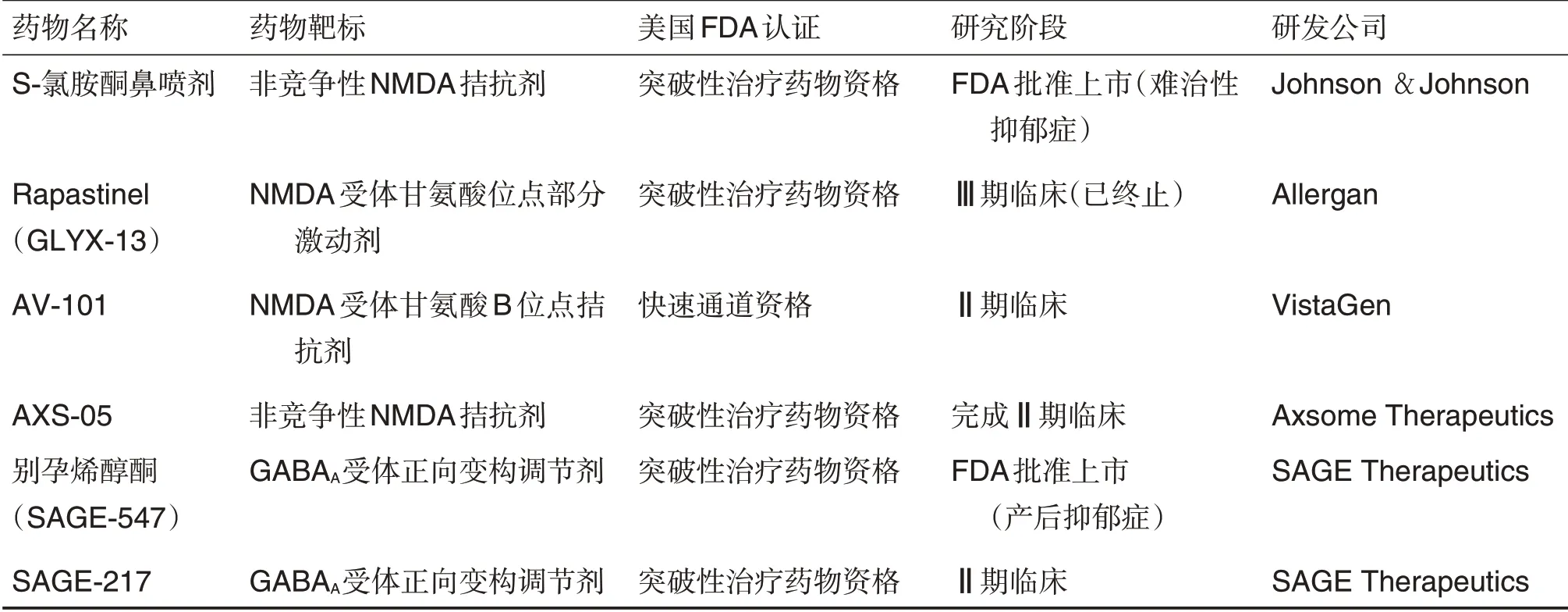
表1 近年获得美国FDA突破性药物或快速通道资格的主要抗抑郁新药
关于Ket 快速抗抑郁作用机制有不同假说,最受关注的是“去抑制假说”。该观点首次提出是由于发现Ket(整体给药30 min后)快速增加啮齿动物PFC 胞外Glu 水平,且该作用仅在低剂量Ket 给药时才出现[32,45]。研究表明,低剂量Ket 选择性阻断GABA 中间神经上的NMDA受体,解除了GABA 能中间神经元对Glu 能锥体神经元的抑制作用,导致Glu突释;进而引起BDNF快速释放(而非BDNF基因表达),通过激活BDNF-mTOR通路快速增强锥体神经元的突触数量和功能,这可能代表了快速起效抗抑郁药独特机制[32,45]。该发现也使mTORC1成为诱人的药物靶点,Navitor生物公司研发的NV-5138通过与sestrin 结合激活mTORC1。单次口服NV-5138 增强mPFC 中mTORC1 通路功能,具有快速抗抑郁活性且可持续7 d[32,46]。此外,单次给予NV-5138 增加mPFC 突触数量功能及突触蛋白水平,在这一过程中BDNF 是必需的。研究证实,BDNF在Ket快速起效中发挥了重要作用,Ket的抗抑郁作用在条件性敲除BDNF 小鼠上消失了,且PFC注射BDNF功能性阻断抗体可阻断Ket抗抑郁作用[47-48]。因此,BDNF 释放是激活突触蛋白合成的关键环节。
3.2 GABAA受体正向变构调节剂
Glu-GABA信号通路稳态的异常被认为介导许多神经精神病。免疫组化分析揭示抑郁患者PFC中各类GABA能中间神经元减少[49],而GABA功能缺失也确实导致“抑郁样”行为[50],提示GABA中间神经元在抑郁症患病机制中起关键作用,Glu-GABA稳态可能与GABA 中间神经元变化有关[32]。近年来,一个令人振奋研究结果是静脉注射神经类固醇四氢孕酮(SAGE-547,即别孕烯醇酮)在产后抑郁症(postpartum depression,PPD)女性中具有快速抗抑郁作用[51]。四氢孕酮是突触及触外GABAA受体的正向变构调节剂,尤其是其通过与δ 亚基调节GABA 中间神经元的点燃[52]。PPD 与分娩时四氢孕酮骤降有关,而在孕期则孕酮源性神经类固醇非常高[32,52]。据报道,敲除δ 亚基的小鼠出现抑郁样行为且产后期有异常的母育行为[53]。临床研究报道,持续iv 别孕烯醇酮在24~48 h 对重度PPD 女性产生抗抑郁疗效[51]。近日别孕烯醇酮已获得FDA 批准上市。此外,Sage 公司研发了可口服的四氢孕酮类似化合物SAGE-217,对男女重度抑郁患者都有疗效。SAGE-217 已获突破性治疗资格(表1),很快将进入Ⅲ期临床[54-55]。
由于四氢孕酮还有抗炎[56]、促进睡眠[57]、保护脑损伤[56]和促进神经元再生[58]等重要作用,因此可能用于治疗抑郁症的共生疾病(即四氢孕酮降低),包括创伤后应激障碍(posttraumatic stress disorder,PTSD)、慢性痛、神经创伤、酒精依赖、烟碱依赖和PPD等。目前,四氢孕酮相关的系列疗法正在被研究用于治疗PTSD,其中包括低剂量SSRI(所用剂量可增加脑内四氢孕酮水平,但又不足以阻断5-HT重摄取)、四氢孕酮前体(如孕烯醇酮)以及其他增加GABA能的神经类固醇药物等[59-60]。
3.3 非单胺策略新药:TSPO配体YL-IPA08
事实上,脑内源性四氢孕酮可由18 ku 转位蛋白(translocator protein 18 ku,TSPO)介导的方式在脑内合成。脑内TSPO 主要位于胶质细胞线粒体外膜上,是介导脑内胆固醇进入线粒体和合成神经类固醇(包括四氢孕酮)的重要蛋白。神经类固醇与GABAA受体上相应位点结合诱导该受体构象发生改变,调节中枢兴奋和HPA轴功能[56,61]。本实验室采用TSPO 敲除动物和病毒过表达研究发现,TSPO-/-与TSPO+/+小鼠虽对慢性应激具有相同的敏感性(即应激后抑郁-认知行为表型相同),但令人惊喜的是,TSPO选择性激活剂AC-5216 在野生型小鼠上快速(2~3 d)产生抗焦虑、抗抑郁和促认知作用,且快速增强突触可塑性是其重要机制,而在敲除小鼠上无此作用,明确了TSPO 的药理学靶标价值。此外,本实验研究还发现TSPO 通过对抗炎性损伤而增强海马神经细胞再生[62]。这些研究为基于胶质细胞的精神疾病治疗理论提供了新思路。进而,我们首次发现TSPO 是PTSD 的潜在重要靶标,TSPO 及其介导的四氢孕酮释放是抗PTSD、抗焦虑-抑郁行为的重要环节[63-68]。这些提示,通过调节胶质细胞增强海马神经元再生与突触可塑性,可能代表着不同于现有药物的全新机制。
基于此,本实验室设计筛选得到的1.1 类抗焦虑、抗抑郁候选新药YL-IPA08,并获系列专利授权;与一线SSRI类药物相比,YL-IPA08 具有快速抗焦虑-抑郁效应、兼可增强认知,且无镇静催眠、肌松、运动平衡障碍和认知损伤等缺陷,表现出良好的效应特点与成药性[63-67]。该化合物的发现,提示GABAA受体为代表的非单胺靶点在抑郁症发生和治疗中的重要作用。
综上所述,脑内Glu/GABA 间的平衡稳态兴奋/抑制(excitation/inhibition,E/I 平衡)在快速突触可塑性调节中发挥了重要作用;我们前期研究发现,YL-0919 和YL-IPA08 在慢性应激模型上3 d 均可快速产生抗抑郁作用,同时增强突触数量和树突复杂性,它们都涉及GABAA受体的功能以及E/I 平衡稳态调节,提示GABA中间神经元可能是单胺能药物(如YL-0919)和非单胺能药物(如YL-IPA08)快速起效抗抑郁的重要汇聚点,还可能介导着锥体神经元兴奋性的调控并最终快速达成E/I 平衡,这或许是快速抗抑郁的重要机制。
4 单胺-非单胺(即5-HT-Glu/GABA)长反馈神经环路候选假说的提出
神经生物学研究表明,PFC 和DR 之间存在神经长投射方式的解剖学联系[69],即DR的5-HT神经元长投射至PFC(还包括海马和杏仁核等),调节GABA 中间神经元和Glu 锥体神经元兴奋性;而PFC 的锥体神经元长投射至DR 调节5-HT 神经元活性。研究证实,5-HT 在PFC 的释放依赖于兴奋性Glu 转导的激活,而PFC 投射到DR 的神经纤维可抑制应激行为[70-71]。PFC的Glu能神经元优先靶向激活DR 的5-HT 能神经元,并激活DR 的5-HT 神经元是依赖谷氨酸释放的。另外的研究发现,DR 中存在许多GABA 中间神经元,并且存在GABAA受体,激活该受体可抑制5-HT释放[72]。
结合本实验室和国外同行研究,我们认为单胺能靶标和非单胺能靶标均可能是介导抗抑郁快速起效的重要环节。多靶标单胺能新药羟哌吡酮(YL-0919)是全新结构的5-HT1A和5-HT6激动剂及5-HT 重摄取抑制剂,非单胺能新药YL-IPA08 是选择性TSPO 激动剂,通过提高脑内四氢孕酮调控GABA 神经功能;二者的共同点是:①均起效快速且毒副作用低;②均有促认知活性;③均对GABA神经元兴奋性有调控作用。这些为单胺(5-HT)与非单胺(GABA)2 方面研究进一步聚焦,发现完整的快速起效调控环路提供了线索。我们认为,DR和PFC 两脑区中均存在着单胺(5-HT)和非单胺(Glu-GABA系统)交互作用的界面(图3),即:①在DR中,以5-HT神经元为中心的Glu-GABA 调节界面,5-HT 神经元活性受到来自PFC 的锥体神经元长投射(通过AMPA受体)以及来自附近GABA中间神经元(通过GABAA受体)的调节;②在PFC(还包括海马等)中,以Glu-GABA 为中心的5-HT等受体调节界面,Glu锥体神经元和GABA神经元受到来自DR 的5-HT 长投射调节(通过5-HT 受体)。且这2 个脑区之间又组成了单胺能与非单胺能相互衔接的长程反馈神经环路(即单胺-非单胺交互作用环路)。基于此,提出介导快速抗抑郁效应的“单胺(5-HT)-非单胺(Glu/GABA)长反馈神经环路假说”,我们认为该环路的快速启动激活以及持续的正性强化可能是快速抗抑郁的重要机制,这使PFC等脑区快速达到E/I 平衡,并通过BDNF-mTOR 通路等快速增强突触可塑性。

图3 快速起效抗抑郁的5-HT-Glu/GABA长反馈神经环路. 5-HT:5-羟色胺;Glu:谷氨酸;GABA:γ-氨基丁酸;E/I 平衡:兴奋/抑制平衡;SSRI:5-HT重摄取抑制剂;mTOR:哺乳动物西罗莫司(雷帕霉素)靶蛋白;BDNF:脑源性神经营养因子;AMPA:α-氨基-3-羟基-5-甲基-4-异恶唑丙酸受体.
基于该环路,至少应该有5 个快速起效抗抑郁的启动策略:①快速E/I 平衡调节,即通过拮抗M1或NMDA 等受体解除GABA 神经元对Glu 锥体神经元的抑制,或直接激活Glu 锥体神经元(如mGluR2/3拮抗剂、AMPA激动剂),或GABAA受体正向变构调节剂(或反向激动剂)等;如Ket,YL-IPA08和SAGE-217属于该类。②同时调控5-HT神经元活性和E/I 平衡(即同时增强单胺-非单胺环节),即一方面基于5-HT1A和5-HT1B等突触前自身受体拮抗剂(或部分激动剂)使受体快速失敏,促进DR 的5-HT大幅释放;同时通过5-HT1A等突触后受体激活Glu锥体神经元(或解除其抑制),如YL-0919和维拉佐酮。③直接激活mTORC1,导致BDNF-mTOR通路激活,引起锥体神经元突触数量和功能的快速增加,如NV-5138。④刺激脑内BDNF 的快速释放或浓度增加(不是BDNF 基因表达,而是释放)。⑤正向变构调节突触及突触外GABAA受体。
SSRI 等之所以起效缓慢的机制可能是不能快速实现E/I平衡,即提高PFC的5-HT水平后,通过系列5-HT受体(包括14种亚型)非特异性的、间接调节E/I 稳态,而这个适应性调节过程需要较长时间重新达到E/I 平衡;我们最近的研究也发现,氟西汀单次给药并不能直接兴奋PFC 锥体神经元。而YL-0919 和Ket 一样,单次给药可激活Glu 锥体神经元(通过5-HT1A受体解除GABA 神经元抑制作用),并导致BDNF 快速释放。另外,SSRI 可增加BDNF基因表达(而非脑内释放),这需要2~3周时间。这些提示非单胺系统调控(E/I 平衡)可能是快速起效的主要启动环节。
基于该候选假说,我们认为脑内不存在排除单胺(如5-HT)之外的非单胺机制,二者是相互偶联的,组成了正性强化长反馈环路,导致该环路的快速激活的单胺和(或)非单胺靶标策略可能是未来重要方向,但还有一些问题需要深入研究,包括①快速E/I 平衡的精细调控机制是什么?②DR 中5-HT 神经元激活和失敏的机制;③该环路是否存在男女敏感性的差异;④该环路与人体其他系统(如肠道菌群)的关系等等。相信相关深入研究将为阐明快速治疗抑郁症的脑机制奠定基础。
5 结语
抗抑郁药发展的60 余年总体上是新药物-新理论循环反复发展的过程,丙咪嗪等药物的诞生催生了经典单胺假说,并指导了现有主要一线药物的研发,随着新药发展,现代单胺策略使更快速、更低毒副作用成为可能,而今年S-Ket 等快速起效抗抑郁药物的上市为非单胺理论(策略)开辟了广阔前景。通过总结本实验室新药研究和国内外进展,作者提出介导快速抗抑郁效应的“单胺(5-HT)-非单胺(Glu/GABA)长反馈神经环路”候选假说,以及基于该假说的5 方面的潜在研发策略,希望这些策略为发展未来新一代抗抑郁药提供潜在突破口,为进一步发现快速抗抑郁候选靶标提供有益的借鉴。
致谢:感谢张黎明副研究员和张有志研究员在成文过程中提出的宝贵建议;感谢尹勇玉博士参与文献调研和补充。谨以此文纪念抗抑郁发展的60年历程。
猜你喜欢
物理实验(2019年4期)2019-05-07 03:36:38
中成药(2018年10期)2018-10-26 03:41:22
天然产物研究与开发(2018年6期)2018-07-09 06:01:46
益寿宝典(2017年26期)2017-10-27 09:09:44
家庭医药(2017年7期)2017-07-15 23:30:57
中成药(2017年6期)2017-06-13 07:30:35
家庭医药(2017年13期)2017-03-25 04:36:34
系统工程与电子技术(2016年2期)2016-04-16 05:16:51
医学研究杂志(2015年5期)2015-06-10 06:43:26
中国中医药现代远程教育(2014年23期)2014-03-01 04:33:37
