
陆地棉低世代群体纤维品质QTL定位及候选基因功能注释
2019-07-24 07:16乔文青严根土石建斌王宁张亚林许庆华周红黄群
棉花学报 2019年4期
乔文青,严根土,石建斌,王宁,张亚林,许庆华,周红,黄群
(中国农业科学院棉花研究所/棉花生物学国家重点实验室,河南安阳455000)
全球陆地棉产量占棉花总产的95%以上[1]。陆地棉高产的优势难以掩盖其纤维品质较差的缺陷,因此以高产为基础、提高陆地棉纤维品质的研究是当前重要的研究方向之一。随着分子标记技术的发展,利用分子标记辅助育种来提高陆地棉纤维品质具有重要意义。
棉花纤维品质性状属于数量性状,具有复杂的遗传背景且易受环境影响[2-4]。传统的分子标记如简单重复序列标记(Simple sequence repeats,SSR)虽然具有成本低、可获得性及稳定性高等特点而被广泛应用于数量性状位点(Quantitative trait loci,QTLs)分析[5-9],但是利用 SSR 筛选获得的QTLs较少、置信区间较大,难以直接应用于分子辅助育种。单核苷酸多态性(Single nucleotide polymorphism,SNP)作为1种重要的分子标记,能够将同一位点的不同等位基因之间有差异的核苷酸标记出来[10],具有位点丰富、数量多、密度大、可覆盖全基因组、代表性高、稳定性高等特点。研究人员利用SNP标记获得大量高密度遗传图谱[11-15],这些遗传图谱为目标性状QTLs定位奠定了基础,而陆地棉基因组的公布[16-17],则为QTLs基因功能注释及挖掘提供了条件,为寻找与纤维品质有关的基因提供了可能性。尽管遗传图谱构建和QTLs作图方法等不断改进和发展,受QTLs的置信区间相对较大、能够解释的表型变异相对较低、相关序列信息缺少等[18-19]的限制,定位精度和准确度依然有待提高,QTLs定位仍处于基础研究阶段,与生产实践结合不紧密。目前尚未见利用分子标记辅助育种方法育成棉花新品系或新品种的报道。因此,构建有指导意义的可重复利用的作图群体、结合陆地棉基因组信息开展QTLs定位研究、利用高密度遗传图谱做精细定位、深度挖掘基因信息等进行标记辅助育种[20]具有重要意义。
本研究主要利用2个陆地棉品种(系)中棉所49和396289构建F2群体,以高密度遗传图谱[21]为基础对纤维品质性状进行QTLs定位,找出在多环境下的低世代群体中稳定存在的QTLs进行基因功能注释,挖掘与纤维品质有关的基因,以提高育种进度及效率。该研究同时为分子标记辅助育种提供理论支撑,为QTLs精细定位和有关功能基因的发掘提供依据。
1 材料与方法
1.1 试验材料与种植方法
本试验以陆地棉栽培品种(系)中棉所49(简称为“中49”)和396289为亲本配制杂交组合,于2015年夏天在中国农业科学院棉花研究所东场试验地(河南安阳)杂交,同年冬天在海南加代获F2,2016年春天将F2群体种植于河南安阳,随机筛选160个单株于7月份取亲本及单株的幼嫩叶片,于-80℃超低温冰箱中储存。对160个单株做自交,构建F2:3家系,将单株自交铃种子随机分成3份,于2017年分别种植于河南安阳(17AY)、安徽安庆(17AQ)以及新疆阿拉尔(17ALE),均采用单行区种植。其中:17AY行长3 m,行间距0.8 m,株距0.25 m;17AQ行长5 m,行间距1.2 m,株距0.5 m;17ALE行长5 m,行间距0.6 m,株距0.1 m。
1.2 DNA提取及分子检测
取中49和 396289及其构建的F2群体的160个单株的幼嫩新叶,采用改良的CTAB法[22]提取DNA。利用亲本重测序和160个子代SLAF简化基因组测序技术(Specific-locus amplified fragment sequencing,SLAF-seq[23])、HighMap[24]软件,对F2遗传分离群体开发高密度分子标记并构建遗传图谱[21]。
1.3 表型数据的获得与分析
随机摘取F2:3家系株行材料的25个正常吐絮棉铃,按照农业农村部棉花纤维品质监督检验测试中心(河南省安阳市)的要求提供材料并委托其检测纤维品质,检测仪器为HVI1000和AFIS pro2。其中:HVI1000检测的指标包括5个,分别为纤维上半部平均长度(纤维长度,FL)、断裂比强度(比强度,FS)、长度整齐度指数(整齐度,FU)、马克隆值(马克隆值,Mic)和断裂伸长率(伸长率,FE);AFIS pro2检测的指标包括纤维细度(细度,Fin)和纤维成熟度(成熟度,Mat)。 品质指标后括号中依次为文中简称和QTLs命名中性状对应的字母标记。
表型数据利用MS Excel 2007统计最大值、最小值、极差、平均值、偏度、峰度等,使用SAS9.2进行方差分析。
1.4 QTLs定位方法
QTLs定位使用软件MapQTL6,利用复合区间作图法,优势对数(Logarithm of odds,LOD)值设为2.5。QTLs的命名规则:(q+性状)+染色体/连锁群+QTL数目[5]。QTLs作用方式用显性度(|D/A|,即显性效应与加性效应的比值的绝对值)表示,|D/A|小于0.20时为加性作用,在0.21~0.80为部分显性作用,在0.81~1.20为显性作用,大于1.20为超显性作用[25]。
1.5 基因功能注释
确定一致性QTLs后,提取亲本QTLs区域的序列信息,进行开放阅读框(Open reading frame,ORF)预测,获得基因信息,同时获取双亲间的非同义突变基因,作为最终的候选基因。应用BLAST软件对置信区间的编码基因通过直系同 源 基 因 簇(Clusters of orthologous groups,COG)、基因本体(Gene ontology,GO)和京都基因与基因组百科全书(Kyoto Encyclopedia of Genes and Genomes,KEGG)等数据库做深度注释。其中:COG数据库可对候选基因产物做直系同源分类;GO数据库从生物过程、分子功能和细胞组分3个方面对候选基因做富集分析;KEGG数据库用于基因的代谢通路分析。
2 结果与分析
2.1 纤维品质表型数据基本统计
在3个F2:3家系中,纤维品质相关性状偏度绝对值均小于1,表型值近似符合正态分布(表 1)。
纤维长度:同一环境下中49长于396289,且二者在不同的环境中均具有极显著差异;整齐度:同一环境下中49高于396289,但不同环境中两者的显著性差异程度不同;比强度:17AY和17AQ的中49极显著高于396289,而17ALE中二者则无显著差异;马克隆值、伸长率、细度:17AY和17AQ的中49和396289有极显著差异,17ALE中则无显著差异;成熟度:中49与396289在17AQ中有显著差异,在17AY和17ALE中无显著差异。
在F2:3家系中,除比强度在17AY和17AQ中为单向超亲分离(F2:3家系中比强度的最大值大于/小于亲本的最大值,家系的最小值大于/小于亲本的最小值)、在17ALE中为双向超亲分离(F2:3家系中的最大值大于亲本的最大值,最小值小于亲本的最小值)之外,其余性状在不同环境均为不同程度双向超亲分布。
2.2 纤维品质性状的相关分析
通过纤维品质相关分析发现,不同环境中纤维品质间的相关性可能不同,但总的来说:纤维长度与比强度、伸长率呈极显著正相关,与细度呈极显著负相关;整齐度与伸长率呈显著正相关;比强度与伸长率呈极显著正相关,与马克隆值、细度呈极显著负相关;马克隆值与细度、成熟度呈极显著正相关;伸长率与细度呈显著负相关;细度与成熟度呈极显著正相关(表2)。
2.3 纤维品质QTLs定位
不同性状的QTLs的统计列于表3。
纤维长度:共获得25个与纤维长度有关的QTLs, 分布于 A03、A04、A07等 8条染色体上,能够解释7%~13.6%的表型变异。17AY、17ALE和17AQ检测到的QTLs分别为2、11和14个,其中qFL-A03-1在3个环境中均能检测到,属于稳定QTL,其加性效应为正,有利等位基因来自父本中49,具有正向部分显性作用。
整齐度:共获得19个与整齐度有关的QTLs,分布于 A02、A03、A04 等 11 条染色体上,能够解释6.9%~16.2%的表型变异。17AY、17ALE和17AQ检测到的QTLs分别为7、8和5个,其中qFU-A04-1在2个环境(17ALE和17AQ)可检测到,属于稳定QTL,其加性效应为正,有利等位基因来自父本中49,具有正向超显性作用。
比强度:共获得25个与比强度有关的QTLs, 分布于 A03、A04、A07等 7条染色体上,能够解释7%~11.5%的表型变异。17AY、17ALE和17AQ检测到的QTLs分别为9、1和15个。
马克隆值:共获得26个与马克隆值有关的QTLs, 分布于 A03、A04、A11等 8条染色体上,能够解释7.1%~16.4%的表型变异。17AY、17ALE和17AQ检测到的QTLs分别为11、7和11 个 ,qMic-A11-1、qMic-D03-1 和 qMic-D12-2均能在17AY和17ALE环境中检测到,属于稳定QTLs,其中qMic-A11-1的加性效应为正,有利等位基因来自父本中49,具有正向超显性作用;qMic-D03-1的加性效应为负,有利等位基因来自母本396289,在17ALE和17AY中分别具有负向显性作用和负向部分显性作用;qMic-D12-2的加性效应在不同环境中有差异,其有利等位基因来自不同的亲本,而QTLs的作用同样表现为不同的方向,但都具有超显性作用。

表1 不同环境中纤维品质相关性状的基本统计Table 1 Basic statistics of traits related to fiber quality traits in different environments
伸长率:共获得19个与伸长率有关的QTLs, 分布于 A03、A04、A08等 6条染色体上,能够解释7.3%~13.3%的表型变异。17AY、17ALE和17AQ检测到的 QTLs分别为 6、1和14个,其中qFE-A04-1和qFE-A04-2在17AY和17AQ环境中均能检测到。qFE-A04-1的加性效应为正,有利等位基因来自父本中49,在17AY和17AQ中分别具有正向超显性和正向显性作用;qFE-A04-2的加性效应同样为正,具有正向显性作用。

表2 不同环境下纤维品质性状的相关分析Table 2 Correlation analysis of fiber quality traits in different environments
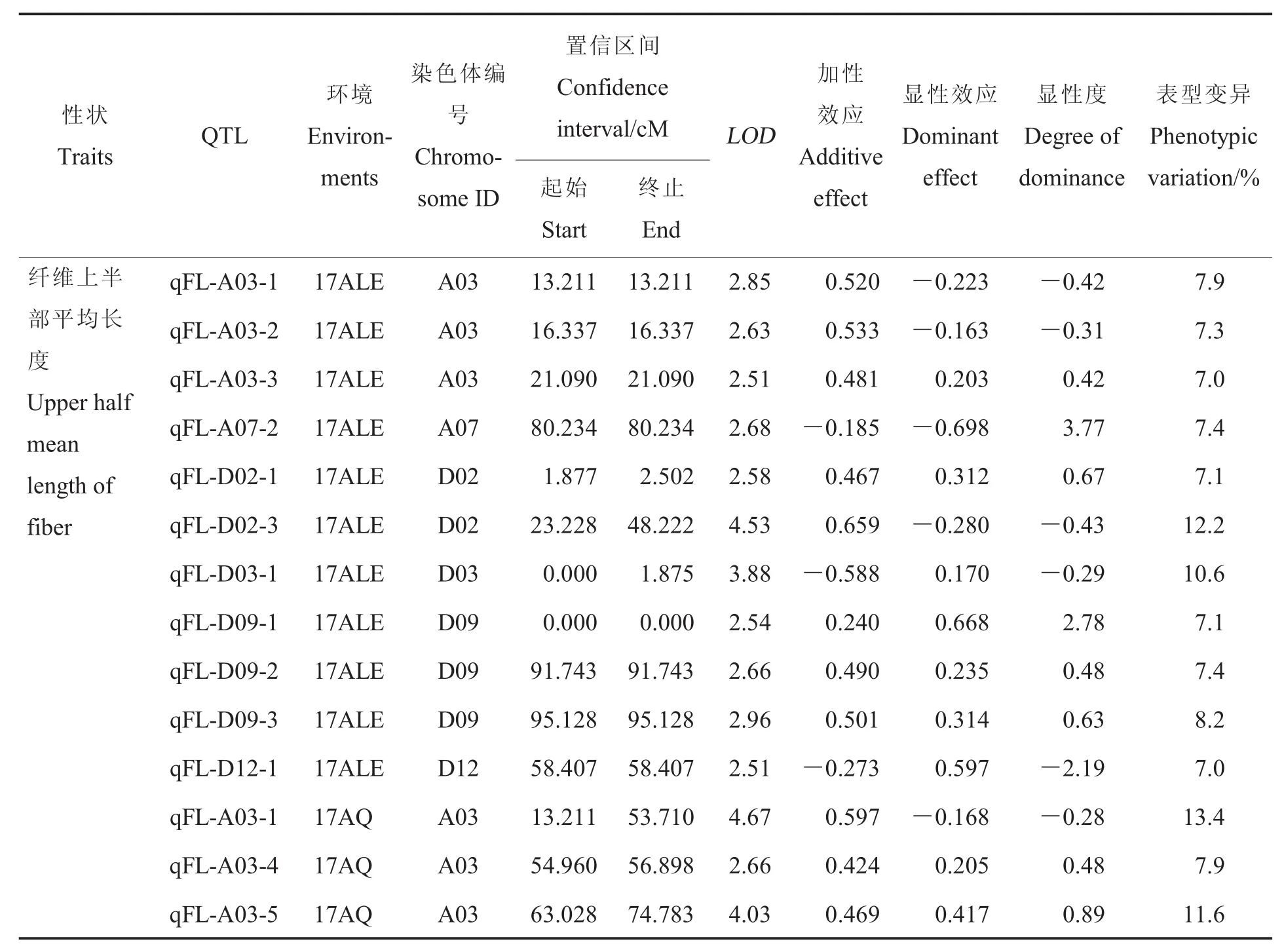
表3 纤维品质相关性状的QTLTable 3 QTLs for fiber quality traits

表3(续)Table 3 (Continued)

表3(续)Table 3 (Continued)

表3(续)Table 3 (Continued)
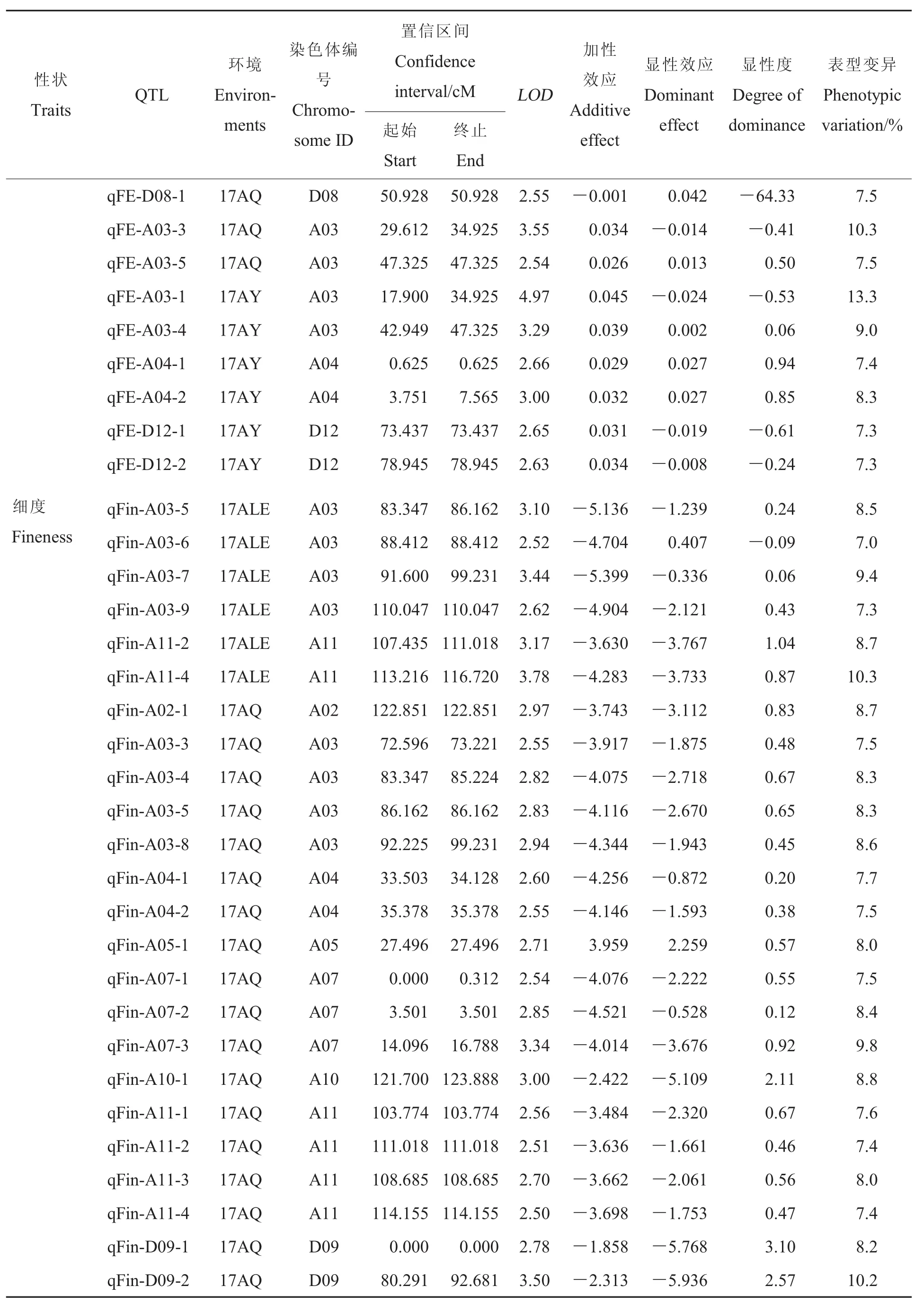
表3(续)Table 3 (Continued)

表3(续)Table 3(Continued)
细度:共获得28个与细度有关的QTLs,分布于A02、A03、A04等11条染色体上,能够解释7%~10.3%的表型变异。17AY、17ALE和17AQ检测到的 QTLs分别为 9、6和 18个,其中qFin-A11-4在3个环境中均能检测到,qFin-A03-5和qFin-A11-2在17ALE和17AQ环境中均能检测到,而qFin-A11-1在17AY和17AQ环境中能检测到。
成熟度:共获得15个与成熟度有关的QTLs, 分布于 A03、A05、A08等 8条染色体上,能够解释7.1%~11.1%的表型变异。17AY、17ALE和17AQ检测到的QTLs分别为8、6和1个。
2.4 纤维品质QTLs候选区域内的基因功能注释
纤维长度:与纤维长度有关的QTLs置信区间中共有1 434个候选基因,其中1 002个被注释,各QTL中候选基因数从0到274个不等(附表1,印刷版略,电子版参见本刊网站)。在COG分类有513个注释基因,其中有151、75、70、69、62个基因分别与一般功能预测、信号转导机制、转录、翻译后修饰/蛋白质周转/伴侣蛋白、复制/重组/修复等有关。在GO富集中有901个注释基因,其中在细胞组成中,有较多的基因与细胞核、膜的组成部分、质膜等有关;在分子功能中,有较多的基因与ATP结合、金属离子结合、DNA结合有关;在生物过程中,有较多的基因与氧化还原过程、转录调控、DNA模板化、蛋白质磷酸化等有关。稳定QTL qFL-A03-1置信区间中只有1个基因Gh_A03G0097,该基因是膜的组成部分,参与硫代葡萄糖苷生物合成过程、植物细胞壁纤维素代谢过程以及细胞壁果胶代谢过程。在KEGG分析中,524个基因参与96条通路,其中,有 31、16、16、16、16 个基因分别与核糖体、 碳代谢、谷胱甘肽代谢、植物-病原体相互作用、蛋白质在内质网中的加工通路有关等。
整齐度:与整齐度有关的QTLs置信区间中有2 024个候选基因,其中1 330个被注释,各QTL中候选基因数从0到954个不等。在COG分类中有636个注释基因,其中有203、121、115、99、56个基因分别与一般功能预测、转录、复制/重组/修复、信号转导机制、翻译后修饰/蛋白质周转/伴侣蛋白等有关。在GO富集中有1 171个注释基因,在细胞组成中,有较多的基因与细胞核、膜的组成部分、质膜等有关;在分子功能中,有较多的基因与ATP结合、金属离子结合、DNA结合等有关;在生物过程中,有较多的基因与氧化还原过程、蛋白质磷酸化、转录调控、DNA模板化有关。在KEGG分析中,693个基因参与107 条通路。 其中,有 28、27、24、23、21 个基因分别与核糖体、碳代谢、植物激素信号转导、RNA运输、氧化磷酸化通路有关等。在稳定QTL qFU-A04-1置信区间中未发现候选基因。
比强度:与比强度有关的QTLs置信区间中有957个候选基因,其中618个被注释,各QTL中候选基因数目从0到316个不等。在COG分类中有 290 个注释基因,其中有 76、47、39、35、33个基因分别与一般功能预测、转录、信号转导机制、复制/重组/修复、翻译/核糖体结构/生物发生等有关。在GO富集中有562个注释基因,在细胞组成中,有较多的基因与细胞核、膜的组成部分、质膜有关;在分子功能中,有较多的基因与ATP结合、DNA结合、金属离子结合有关;在生物过程中,有较多的基因与氧化还原过程、转录调控、DNA模板化、蛋白质磷酸化有关。在KEGG分析中,321个基因参与94条通路。其中,有 22、14、11、9、8 个基因分别与核糖体、 氨基酸的生物合成、碳代谢、植物激素信号转导、谷胱甘肽代谢通路有关。
马克隆值:与马克隆值有关的QTLs置信区间中有1 454个候选基因,其中990个被注释,各QTL中候选基因数目从0到630个不等。在COG分类中有506个注释基因,其中有143、62、59、58、47个基因分别与一般功能预测、转录、信号转导机制、复制/重组/修复、翻译后修饰/蛋白质周转/伴侣蛋白等有关。在GO富集中有872个注释基因,在细胞组成中,有较多的基因与膜的组成部分、质膜、细胞核有关;在分子功能中,有较多的基因与ATP结合、锌离子结合、金属离子结合有关;在生物过程中,有较多的基因与氧化还原过程、转录调控、DNA模板化、代谢过程有关。在KEGG分析中,518个基因参与96条通路。 其中,37、25、17、16、15 个基因分别与植物激素信号转导、核糖体、碳代谢、内质网中的蛋白质加工、淀粉和蔗糖代谢通路有关等。稳定QTL的qMic-A11-1的置信区间中无候选基因;qMic-D03-1共 有 424个 候 选 基 因(Gh_D03G1014—Gh_D03G1437),其中有注释信息的基因有309个,在COG、GO和KEGG中分别有163、276和173个;qMic-D12-2的置信区间中共有122个候选基因(Gh_D12G1203—Gh_D12G1324),其中有注释信息的基因有87个,在COG、GO和KEGG中分别有49、82和46个。
伸长率:与伸长率有关的QTLs置信区间中有535个候选基因,其中364个被注释,各QTL中候选基因数目从0到215个不等。在COG分类中有 177 个注释基因,其中有 51、28、24、23、17个基因分别与一般功能预测、转录、复制/重组/修复、信号转导机制)、翻译后修饰/蛋白质周转/伴侣蛋白等有关。在GO富集中有335个注释基因,在细胞组成中,有较多的基因与细胞核、膜的组成部分、质膜有关;在分子功能中,有较多的基因与ATP结合、DNA结合、金属离子结合有关;在生物过程中,有较多的基因与氧化还原过程、转录调控,DNA模板化、蛋白质磷酸化有关。在KEGG分析中,208个基因参与72条通路。其中,10、9、9、7、5 个基因分别与碳代谢、植物 -病原体相互作用、核糖体、氨基酸的生物合、戊糖和葡萄糖醛酸相互转化通路有关等。在稳定QTL qFE-A04-1和qFE-A04-1置信区间中未发现候选基因。
细度:与细度有关的QTLs置信区间中有946个候选基因,其中613个被注释,各QTLs中候选基因数目从0到225个不等。在COG分类中有 305 个注释基因, 其中有 86、43、38、29、29个基因分别与一般功能预测、转录、信号转导机制、复制/重组/修复、翻译后修饰/蛋白质周转/伴侣蛋白等有关。在GO富集中有512个注释基因,在细胞组成中,有较多的基因与膜的组成部分、质膜、细胞核有关;在分子功能中,有较多的基因与ATP结合、金属离子结合、核苷酸结合有关;在生物过程中,有较多的基因与氧化还原过程、转录调控,DNA模板化、跨膜运输有关。在KEGG分析中,333个基因参与92条通路。其中,33、11、10、9、8 个基因分别与植物信号转导、氨基酸的生物合成、核糖体、吞噬、氧化磷酸化通路等有关。稳定QTLs qFin-A11-1和qFin-A11-2置信区间中无候选基因,qFin-A11-4的置信区间中14个候选基因(Gh_A11G2846—Gh_A11G2859),其中有注释信息的基因有10个,在COG、GO和KEGG中分别有3、8和6个;qFin-A03-5共有32个候选基因(Gh_A03G0852—Gh_A03G0883),其中有注释信息的基因有22个,在COG、GO和KEGG中分别有11、20和10个。
成熟度:与成熟度有关的QTLs置信区间中有1 728个候选基因,其中1 240个被注释,各QTL中候选基因数目从0到703个不等。在COG分类中有643个注释基因,其中有200、126、112、107、69 个基因分别与一般功能预测、转录、信号转导机制、复制/重组/修复、翻译/核糖体结构/生物发生等有关。在GO富集中有1 092个注释基因,在细胞组成中,有较多的基因与膜的组成部分、细胞核、质膜有关;在分子功能中,有较多的基因与ATP结合、金属离子结合、核糖体的结构成分有关;在生物过程中,有较多的基因与氧化还原过程、转录调控,DNA模板化、蛋白质磷酸化有关。在KEGG分析中,667个基因参与 100 条通路。 其中,53、27、24、22、19 个基因分别与核糖体、植物激素信号转导、碳代谢、氨基酸的生物合成、内质网中的蛋白质加工通路等有关。
3 讨论
3.1 纤维品质各指标间的表型分析
通过对亲本和F2:3家系在多环境下表现研究发现,同一环境中,亲本的成熟度差异不明显,但其他性状有显著差异,这可能说明环境对成熟度影响不大。纤维品质相关性分析表明,性状间的相关性因环境而异,但不同环境中,马克隆值与细度和成熟度间的相关性一致,这可能与马克隆值是细度和成熟度的综合指标有关。基于纺纱工业对纤维品质的要求,本研究结果可为育成纺织工业要求的棉花新材料提供理论依据,同时为同步改良部分纤维品质性状提供理论依据。
3.2 纤维品质相关QTL定位
亲本材料、群体类型和分子标记类型等方面的不同为发现大量QTLs提供可能,同时也有助于发现控制同一性状的稳定QTLs[5-9]。随着高通量测序技术的发展,利用高密度遗传图谱[19,26]对纤维品质进行定位越来越普遍,而前人的研究大多集中在纤维长度、整齐度、比强度、马克隆值和伸长率等5个指标,很少研究细度和成熟度等指标。本研究通过对包括细度和成熟度在内的7个纤维品质性状定位,共获得157个与纤维品质有关的QTLs,其中有99个QTLs的遗传距离在1 cM以下,与前人[3,9,19]的研究相比,QTLs的置信区间明显缩小。qFL-A03-1和qFin-A11-4在3环境中均能检测到,有9个QTLs在其中2个环境中能检测到,这说明了低世代群体中也可能存在稳定QTLs,这些稳定的QTLs对分子标记辅助育种具有重要参考价值。除此之外,有7个性状的QTLs分布在A03上,6个性状的QTLs分布于A04和D02上,5个性状的QTLs分布于A11和D07上,这种多个性状的QTLs集中在同一条染色体呈簇分布的现象,与He等[27]和秦永生等[28]的研究结果相似,这些QTLs簇可能与纤维品质各性状有关, 同时也说明 A03、A04、D02、A11 和D07等染色体极有可能是控制纤维品质性状的关键染色体,可作为纤维品质改良的重点研究内容,在分子标记辅助育种中具有重要参考价值。
3.3 纤维品质QTLs区域内候选基因的功能注释
在7个性状中,共有4 763个注释基因,其中在COG中有2 416个注释基因,分为24类,其中含有基因数较多的类有一般功能预测、转录、信号转导机制、复制/重组/修复、翻译后修饰/蛋白质周转/伴侣蛋白、翻译/核糖体结构/生物发生等,这些基因可能与纤维发育有关,影响纤维品质。GO富集中有4 188个注释基因,其中,在生物过程中含有基因最多的3个亚类包括氧化还原过程、转录调控、DNA模板化;在细胞组成中含有最多基因的3个亚类是细胞核、膜的组成部分、质膜;在分子功能分类中还有基因最多的3个亚类是ATP结合、金属离子结合、核糖体的结构成分;该结果与各性状的GO分类结果类似。Qin等[29]和Tang等[30]研究表明,活性氧在纤维发育中起关键作用,在本研究中与纤维长度有关的基因Gh_A03G0483参与对活性氧的反应,与整齐度有关的基因Gh_A07G0233参与对活性氧的反应,Gh_A11G2849参与活性氧代谢过程的调控;与比强度有关的基因Gh_A11G2849参与活性氧代谢过程的调控;与马克隆值有关的基因Gh_D03G1055参与对活性氧的反应;与细度有关的基因Gh_A11G2849参与活性氧代谢过程的调控。上述基因可能是调控纤维品质的重要基因,其作用机理有待进一步研究。在KEGG通路中有2 521个注释基因参与121个通路,其中核糖体、植物激素信号转导、碳代谢通路是参与基因数最多的通路。一些研究表明类黄酮途径可能参与纤维的发育过程[31-33]。同样有研究表明植物激素信号转导途径对纤维发育有影响[34]。上述与纤维发育有关的通路,在本研究中均有注释到,因此与这些通路相关的基因可能参与纤维发育的过程,有可能影响纤维品质,但其作用机理有待进一步研究。
4 结论
本研究以高密度遗传图谱为基础,对纤维品质性状QTLs进行定位及候选基因功能注释,主要结论如下:(1)纤维品质各性状的表型分析发现,纤维长度与比强度和伸长率呈极显著正相关、与细度呈极显著负相关,纤维强度与伸长率呈极显著正相关、与马克隆值和细度呈极显著负相关,马克隆值与细度和成熟度呈极显著正相关,该结果不仅为筛选适宜纺织工业的新材料提供理论依据,而且为同步改良部分纤维品质性状提供理论依据。(2)本研究通过对包括细度和成熟度在内的7个纤维品质性状进行QTLs定位,获得157个QTLs,分布于20条染色体上,有的染色体上分布有控制多个性状的QTLs,如:A03、A04和D02等,这些染色体极有可能是控制纤维品质性状的关键染色体。除此之外,获得了11个稳定的QTLs,其中qFL-A03-1和qFin-A11-4在3个环境中重复出现,另有9个QTLs在2个环境中重复出现。(3)在 11个稳定 QTLs中,qFL-A03-1、qMic-D03-1、qMic-D12-2、qFin-A11-4和qFin-A03-5上共有593个候选基因,其中有429个有注释信息,可通过进一步的筛选来验证是否是控制纤维品质的关键基因。
附表1:QTL候选基因在COG、GO和KEGG数据库的基因功能注释信息(见期刊网站)
猜你喜欢
江西师范大学学报(自然科学版)(2022年4期)2022-10-18
昆钢科技(2022年2期)2022-07-08
湖南农业大学学报(自然科学版)(2021年4期)2021-08-14
云南医药(2021年3期)2021-07-21
纺织检测与标准(2021年1期)2021-04-07
内江师范学院学报(2021年2期)2021-03-03
安徽工业大学学报(自然科学版)(2020年4期)2020-12-26
商品与质量(2020年23期)2020-11-26
计算机与网络(2020年13期)2020-07-29
心理技术与应用(2019年5期)2019-05-24
