
丹参中有效成分联合提取工艺的优化
2019-07-23 11:42:54林大专张义英王俊儒
中成药 2019年6期
林大专, 张义英, 王俊儒
(1.长春医学高等专科学校,吉林 长春130031;2.西北农林科技大学理学院,陕西 杨凌712100)
丹参为唇形科植物丹参Salvia miltiorrhiza Bge.的干燥根及根茎,主要有效成分为水溶性的酚酸类和脂溶性的丹参酮类,药理活性广泛,在治疗心血管疾病、肝硬化、溃疡、肿廇、新生儿缺氧性脑病等具有显著疗效[1]。上述2种成分在同一工艺中不易兼得,但多种新技术联合、多种成分标准控制的生产工艺是近几年发展趋势,后者一般采取传统的回流提取,适合工业化生产[2];也有用超声提取的,提取率较高[3],一般作为辅助技术;还有超临界萃取法,但成本较高[4],而前者常采用水煮醇沉法。目前,尚无对丹参中脂溶性、水溶性成分同时提取的报道,故建立联合提取工艺很有必要,这样既可降低成本,又能提高有效成分利用率,故本实验采用正交试验优化丹参中有效成分(酚酸类和丹参醌类) 联合提取(回流提取为主,超声提取为辅) 工艺,以期为这些成分开发利用提供参考。
1 材料
丹参购于安国市昌达中药材饮片有限公司,经长春医学高等专科学校生药教研室王月珍老师鉴定为正品,符合2015 年版《中国药典》 一部规定。丹参酮ⅡA对照品(成都普思生物科技股份有限公司,批号Q0070291,50 mg,含有量≥99%);丹酚酸B 对照品(北京百欧博伟生物技术有限公司,批号151219,20 mg/支,含有量≥98%);
Agilent 1200 高效液相色谱仪,配置Agilent 1200 可变波长检测器(VWD)、Agilent 1200 数据站、P/N-7725i 手动进样器、G1354A 四元梯度泵、G1316A 柱温箱 (美国Agilent 公司);BS224S 电子天平[赛多利斯科学仪器(北京) 有限公司];KQ-2200E 超声波清洗器(昆山市超声仪器有限公司);HH-2 恒温水浴锅(金坛市新航仪器厂)。
甲醇为色谱纯(上海兴科高淳溶剂有限公司,批号021260703);乙腈为色谱纯(美国Honeywell 公司,批号10071743); 无 水 乙 醇 (批 号2015130)、 甲 酸 (批 号20040315) 为分析纯(北京化工厂)。
2 方法
2.1 联合提取 参考文献[5] 发现,70%乙醇提取效果最好,同时结合脂溶性或水溶性单一成分采用超声提取。取丹参约2.0 g,先用6 倍量70%乙醇超声提取2 次,每次30 min,温度50 ℃,合并提取液,然后药渣进行回流提取,最后将超声、回流提取液合并,定容于100 mL 量瓶中。以乙醇体积分数(A)、料液比(B)、提取时间(C)为影响因素,正交试验优化提取工艺,因素水平见表1,结果见图1~2。

表1 因素水平
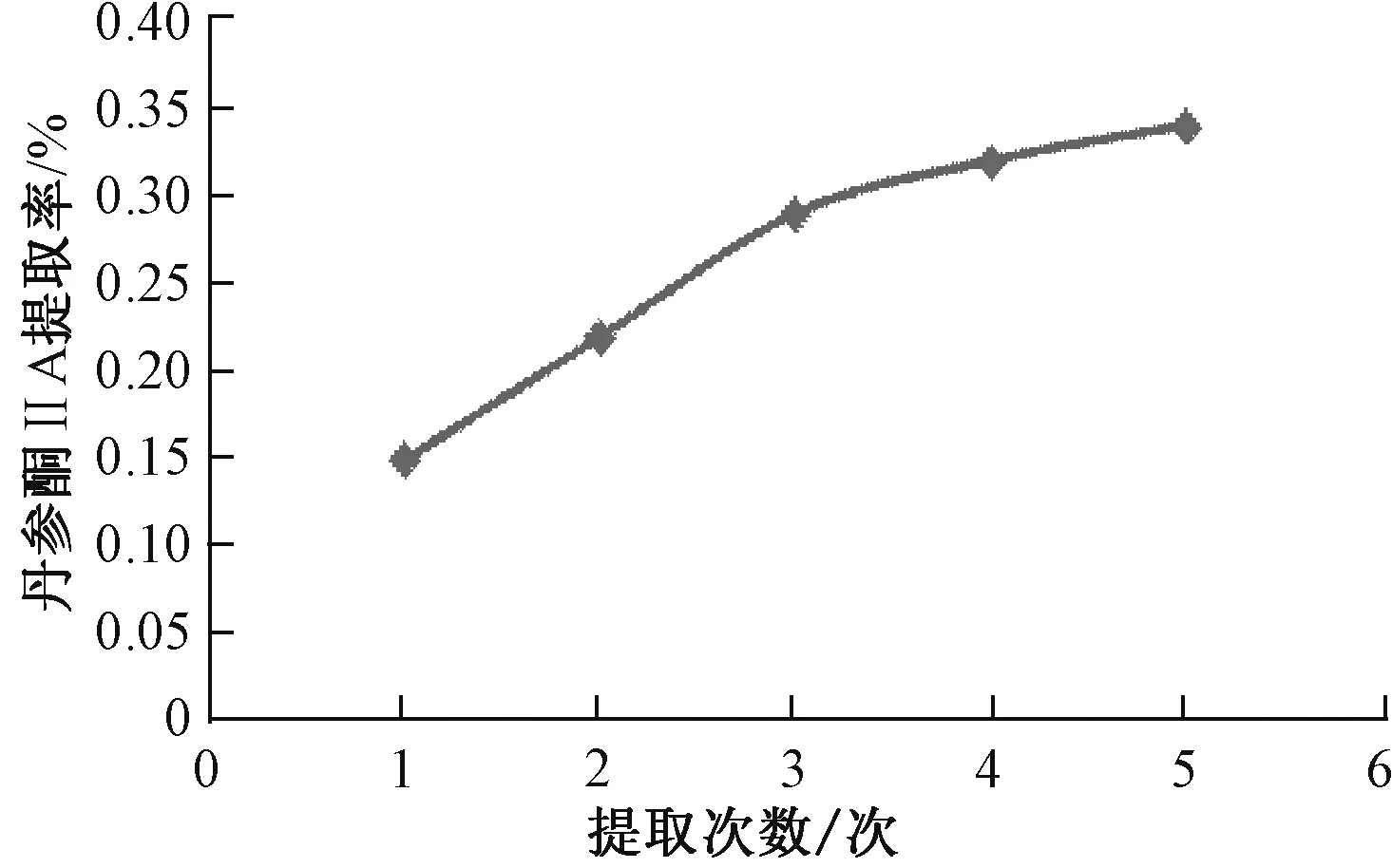
图1 提取次数对丹参酮ⅡA 提取率的影响
2.2 丹参酮ⅡA含有量测定
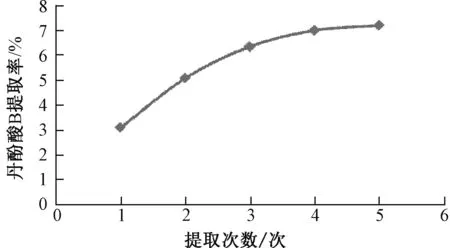
图2 提取次数对丹酚酸B 提取率的影响
2.2.1 色谱条件 Agilent TC-C18色谱柱(250 mm×4.6 mm,5 μm);流动相甲醇-水(80 ∶20);检测波长270 nm;体积流量1.0 mL/min;柱温30 ℃;进样量20 μL。
2.2.2 对照品溶液制备 精密称取丹参酮ⅡA对照品0.005 2 g,甲醇溶解定容至50 mL,即得(104 μg/mL)。
2.2.3 线性关系考察 精密吸取对照品溶液0.5、1.0、1.5、2.0、2.5、3.0 mL 至10 mL 量瓶中,甲醇稀释至刻度,精密量取20 μL,在“2.2.1” 项色谱条件下进样测定。以峰面积(Y) 对溶液质量浓度(X) 进行回归,得方程为Y=164.4X+33.76(r=0.999 3),在5.2~31.2 μg/mL范围内线性关系良好。
2.2.4 精密度试验 精密量取对照品溶液1.5 mL 于10 mL量瓶中,甲醇定容至10 mL,平行6 份,在“2.2.1” 项色谱条件下进样测定, 测得丹参酮ⅡA峰面积RSD 为1.261%,表明仪器精密度良好。
2.2.5 稳定性试验 称取丹参10 g 于250 mL 锥形瓶中,6倍量70%乙醇提取2 次,提取液浓缩至50 mL,精密量取15 mL 于25 mL 量瓶中,70%乙醇定容,摇匀,取6 份,每隔40 min 在“2.2.1” 项色谱条件下进样测定至4 h,测得丹参酮ⅡA峰面积RSD 为0.858%,表明溶液在4 h 内稳定性良好。
2.2.6 重复性试验 按“2.2.5” 项下方法制备提取液,平行6 次,在“2.2.1” 项色谱条件下进样测定,测得丹参酮ⅡA含有量RSD 为1.694%,表明该方法重复性良好。
2.2.7 加样回收率试验 精密量取提取液4.0 mL 于10 mL量瓶中,共9 份,加入丹参酮ⅡA对照品溶液0.5、1.0、1.5 mL,分别配制成高、中、低质量浓度,各3 份,70%乙醇定容,在“2.2.1” 项色谱条件下进样测定,计算回收率。结果,丹参酮ⅡA平均加样回收率为100.75%,RSD为1.07%。
2.3 丹酚酸B 含有量测定
2.3.1 色谱条件 Agilent TC-C18色谱柱(250 mm×4.6 mm,5 μm);流动相甲醇-乙腈-甲酸-水(25 ∶15 ∶1 ∶59);检测波长286 nm;体积流量1.0 mL/min;柱温20 ℃;进样量20 μL。
2.3.2 对照品溶液制备 精密称取丹酚酸B 对照品0.025 0 g,80%甲醇溶解,转移至25 mL 量瓶中,即得(1 mg/mL)。
2.3.3 线性关系考察 精密量取对照品溶液0.5、1.0、1.5、2.0、2.5、3.0、3.5 mL 至5 mL 量瓶中,80%甲醇定容至5 mL,在“2.3.1” 项色谱条件下进样测定。以峰面积(Y) 对溶液质量浓度(X) 进行回归,得方程为Y=16 780X-669.89(r=0.999 4),在0.1~0.7 mg/mL 范围内线性关系良好。
2.3.4 精密度试验 精密量取对照品溶液1.0 mL 至5 mL量瓶中,80%甲醇稀释至刻度,摇匀。精密量取6 份,在“2.3.1” 项色谱条件下进样测定,测得丹酚酸B 峰面积RSD 为0.473%,表明仪器精密度良好。
2.3.5 稳定性试验 精密称取药材约2 g 于100 mL 具塞锥形瓶中,加入10 倍量50%乙醇,40 ℃下超声提取3 次,每次30 min,提取液过滤合并,定容至100 mL 量瓶中,取6 份,每隔40 min 在“2.3.1” 项色谱条件下进样测定至4 h,测得丹酚酸B 峰面积RSD 为0.617%,表明该方法稳定性良好。
2.3.6 重复性试验 按“2.3.5” 项下方法制备提取液,平行6 次,在“2.3.1” 项色谱条件下进样测定,测得丹酚酸B 含有量RSD 为0.844%,表明该方法重复性良好。
2.3.7 加样回收率试验 精密量取“2.3.5” 项下提取液5 mL 于50 mL 量瓶中,50%乙醇稀释至刻度,精密量取2.0 mL 于5 mL 量瓶中,共9 份,加入对照品溶液0.5、0.7、0.9 mL,配制成高、中、低质量浓度各3 份,50%乙醇定容,在“2.3.1” 项色谱条件下进样测定,计算回收率。结果,丹酚酸B 平均加样回收率为100.56%,RSD为1.66%。
3 结果
综合评价[6-7]是运用数学方法(包括数理统计方法)对一个复杂系统的多个指标信息进行分析处理,以得到多个指标之间的优劣的评价方法。由于本实验是通过联合提取对丹参中酚酸类和丹参酮类同时进行提取,故丹酚酸B和丹参酮ⅡA都是重要的考察指标,两者权重系数均为0.5,再进行加权求和,综合评分=0.5×丹酚酸B 含有量/最大值+0.5×丹参酮ⅡA含有量/最大值。结果见表2,方差分析见表3。
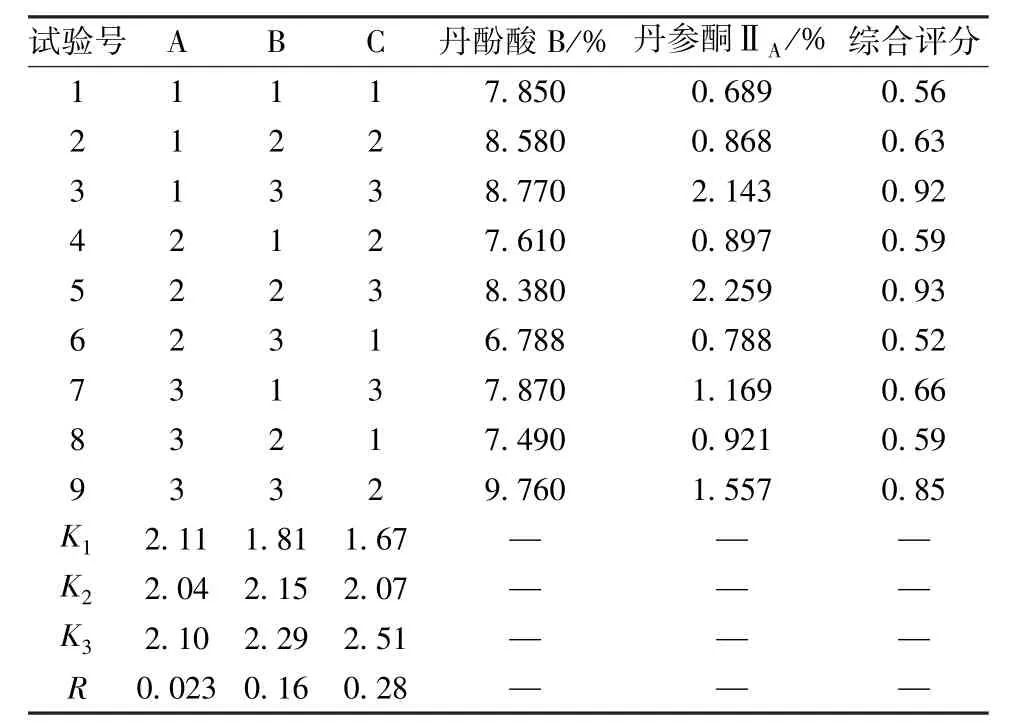
表2 试验设计及结果
由表2 可知,各因素影响程度依次为C(提取时间) >B(料液比) >A(乙醇体积分数)。其中因素A 为K2<K3<K1,因素B 为K1<K2<K3,因素C 为K1<K2<K3;由表3 可知,因素B、C 有显著差异(P<0.05),而因素A 无显著差异(P>0.05),最终确定最优工艺为A1B3C3,即先用6倍量70%乙醇超声提取2 次,每次30 min,提取温度50 ℃,再用30%乙醇回流提取3 次,每次90 min,料液比1 ∶10。根据上述优化工艺进行3 批验证试验,结果见表4,可知工艺稳定可行。
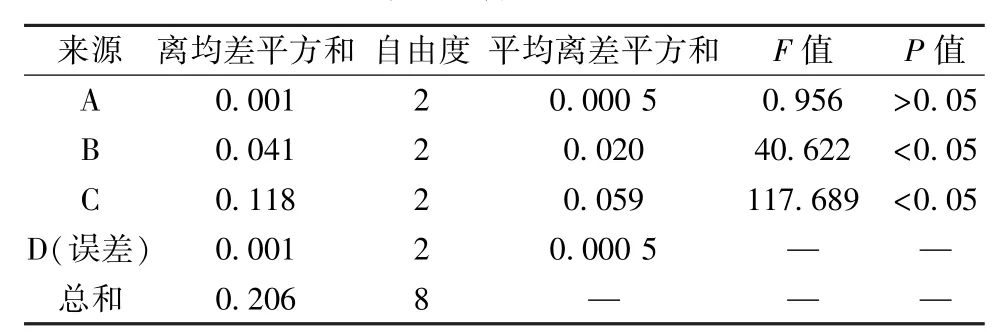
表3 方差分析
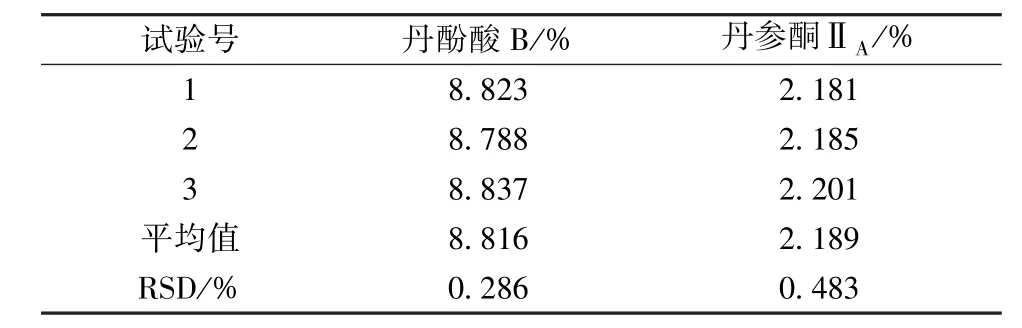
表4 验证试验结果(n=3)
然后,进行单一方法对照平行试验,结果见表5,可知此时丹酚酸B、丹参酮ⅡA含有量明显低于联合提取时。
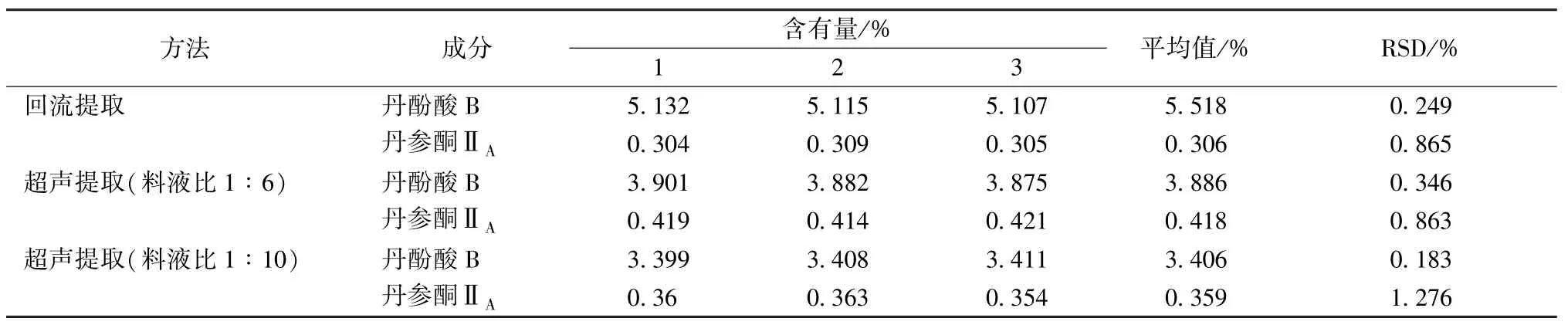
表5 单一方法对照平行试验结果(n=3)
4 讨论
本实验选择提取次数3 次,乙醇体积分数30%、50%、70%,这样有利于丹参酮类、酚酸类成分同时提取。单用回流提取时,乙醇体积分数对丹参酮ⅡA、丹酚酸B 含有量有明显影响;联合提取时,该因素无明显影响,其原因可能是70%乙醇超声提取时已将两者部分提取出,再用不同体积分数乙醇回流提取后即无明显差异。
前期研究[8-14]大多仅以水溶性的酚酸类成分或脂溶性的丹参酮类成分为指标来优化丹参提取工艺,并均采用单一提取方法;本实验同时考虑上述2 种成分,并采用联合提取,发现该方法稳定可行,两者含有量较高,而且有利于质量控制,提高药材利用率,降低生产成本,为实际应用提供参考依据。另外,由于丹参有效成分的提取方法很多,而本实验仅选择超声、回流联合提取,故对其他提取方法的联合效果尚有待于进一步研究。
猜你喜欢
Medicinal Plant(2020年2期)2020-05-14 12:11:26
山东化工(2018年15期)2018-09-20 08:55:34
中成药(2018年3期)2018-05-07 13:34:45
长春中医药大学学报(2017年1期)2017-04-16 05:56:42
首都食品与医药(2015年18期)2015-11-03 05:59:08
电源技术(2015年7期)2015-08-22 08:48:52
电测与仪表(2015年7期)2015-04-09 11:40:30
电测与仪表(2014年9期)2014-04-15 00:27:16
电测与仪表(2014年11期)2014-04-04 09:21:40
中国现代应用药学(2013年4期)2013-03-11 19:13:53
