
基于间隙模式的并发序列模式挖掘算法
2019-07-18 10:45杨梦涛王翠青陈未如
沈阳化工大学学报 2019年2期
杨梦涛, 王翠青, 陈未如
(沈阳化工大学 计算机科学与技术学院, 辽宁 沈阳 110142)
结构关系模式(Structural Relation Patterns,SRPs)是一种后序列模式挖掘[1]的产物,描述了多个序列组成的复杂结构关系.结构关系模式挖掘首先研究的是序列模式之间的关系,然后再把这种关系进一步分解、细化,整合成一种由并发、互斥、重复及串行关系组成的复合结构模式[2].例如,结构关系模式中的并发序列模式实现基于Web浏览中用来挖掘用户浏览记录里的并发关系,根据用户每次浏览的喜好,分析客户习惯,实现基于Web用户行为分析、浏览兴趣的网站设计[3].结构关系模式在蛋白质挖掘中也有重要应用.传统的序列模式挖掘只关注挖掘蛋白质序列中频繁出现的子序列,然而,在蛋白质中一个特殊的功能点往往不是由一个子序列构成,可能是多个基序共同作用,这种结构关系隐藏在蛋白质序列中.所以,可以在传统序列模式的基础上利用结构关系模式挖掘这些子序列之间的结构关系,以便高效地提取生物信息和分析蛋白质功能组成规则.王翠青等人提出的ConSP算法是使用支持向量[4]数据结构存放这些子序列在蛋白质数据库序列中的匹配情况,并进行并发挖掘.实际蛋白质数据集的实验突显了ConSP方法在蛋白质这种数据挖掘中的适用性[5].
生物信息学是由统计学、生物学、计算机学等多门学科构成的一门交叉复杂学科,也是目前生物研究的热点学科之一.生物信息学研究的热点之一就是生物序列模式挖掘.它对识别基因和功能解释、识别非编码区功能元素以及蛋白质序列组成信息的识别等具有重要的指导意义.在生物序列中挖掘频繁模式或者挖掘特定模式是其中两个重要研究领域.1995年,Agrawal和Srikant给出用于交易序列数据库的序列频繁模式挖掘[6]的定义,如Apriori、GSP、PrefixSpan、SPADE等算法.在这些算法基础上,研究者们设计了多个针对生物序列的模式挖掘算法[7].
在序列模式挖掘中有时需要有时间限制约束条件,以保证序列事件或元素之间满足一定的间隔限制.在蛋白质中,通常一个功能点可能是由多个蛋白质基序共同组成.为了有效地发现这些基序之间的关系,Chen-Ming Hsu等人提出WildSpan算法[8].此算法基于Prefixspan算法[9],在此基础上增加了蛋白质氨基酸序列之间的间隙约束.首先挖掘出蛋白质序列中带有固定间隙约束的块,然后通过这些块之间的序列关系来挖掘出它们顺序出现的W-模式.
本文首先提出间隙模式、刚隙模式等概念,试图在通配符间隙约束条件挖掘[10]和One-Off[11]条件的基础上,将间隙体现在模式挖掘结果当中,并提出了基于间隙模式的并发序列模式思想和基于刚隙模式的并发序列模式算法PBcon.与已有的WildSpan算法的W-模式结果集比较,PBcon算法找到了WildSpan算法未发现的模式,且PBcon算法的挖掘效率也有优势.
1 相关概念
1.1 并发关系、并发度及并发序列模式
并发关系、并发度及并发序列模式的定义在参考文献[4]中已有详细的描述.
1.2 间隙模式及刚隙模式
间隙模式:间隙模式是指序列模式中个别位置可以是数据库元素集Σ的任意元素,该位置用通配符x表示,称作间隙.在客户序列数据库中,一个间隙模式被表示为:
GP=a1-x(s1,t1)-a2-x(s2,t2)-…-
x(sn-1,tn-1)-an
(1)
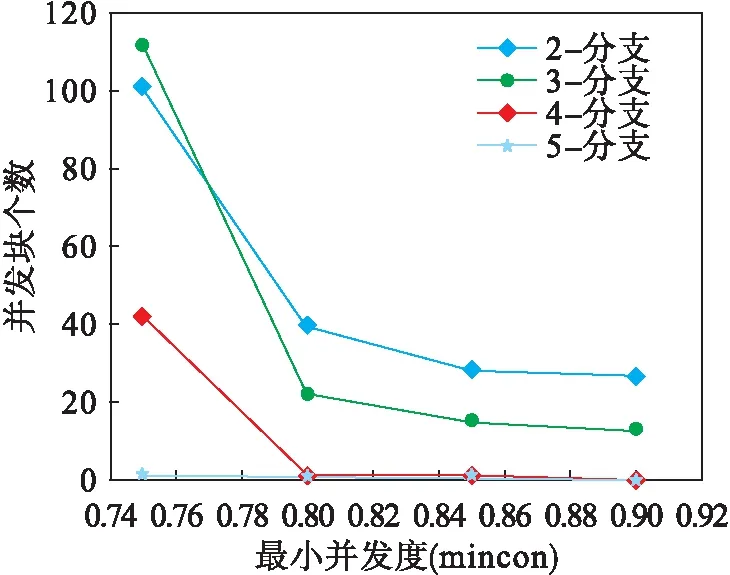
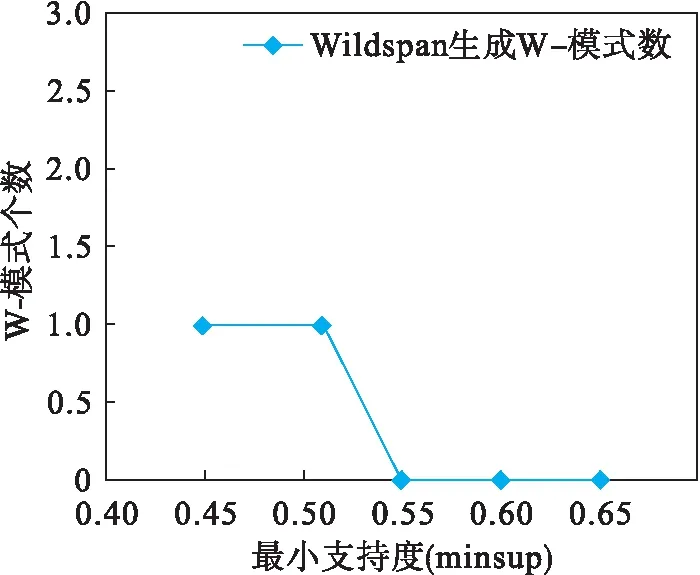
式中,ai是元素,x(si,ti)是元素间的间隙,si和ti是间隙个数下限和上限,即x(si,ti)表示该位置对应si到ti个通配符,其中0≤si≤ti.若si 刚隙模式:刚隙模式是一种特殊的间隙模式,其中只允许存在刚性间隙,表示为 RGP=a1-x(s1)-a2-x(s2)-…- x(sn-1)-an (2) 子间隙模式:从间隙模式p首或尾部删除若干个元素得到新的序列s,并保证s两端不存在通配符区域,则s仍是一个间隙模式,称s为间隙模式p的子间隙模式,p为间隙模式s的超间隙模式. 确型间隙模式、泛型间隙模式:若间隙模式p的通配符区域x(si,ti)的某个通配符置换为元素集Σ的某个确定元素,则可得到一个新的间隙模式s,称s为p的确型间隙模式,p是s的泛型间隙模式. 性质1 间隙模式与其子间隙模式之间存在支持关系,即对于间隙模式s和p,若p∠s有s→p. 性质2 如果一个间隙模式是频繁的,则该间隙模式的子间隙模式也必然是频繁的,该间隙模式的泛型间隙也必然是频繁的. 间隙模式支持量:对于给定的序列数据库SDB,间隙模式a的支持量BSV(a)定义为一个长度为n的二进制向量 (3) 序列的并发间隙模式和并发间隙模式集:若序列数据库的序列s对于间隙模式GPa和GPb的支持分量BSVs(GPa)=BSVs(GPb)=1,则GPa和GPb在该序列中满足并发关系.一般地,所有被序列s所支持的间隙模式c1,c2,…,cm在该序列中满足并发关系,称它们为在序列s上的并发间隙模式,表示为[c1+c2+…+cm]s,构成序列s上的并发间隙模式集{c1,c2,…,cm}. 参考了文献[4]中并发全集和最大并发集的概念,本算法应用了间隙模式集、间隙模式最大集、并发间隙模式最大集等概念,这里不再详细阐述. 输入:序列数据源SDB,最小并发度mincon. 输出:并发间隙模式最大集MCGP. 算法: (1) 在SDB上以mincon为最小支持度挖掘间隙模式,得到间隙模式最大集SetOfGP,并计算其中每个间隙模式的支持量BSV; (2) 令CGP=null,CGPK=SetOfGP=null,k=0; (3) do NewSet=null; k++; for each c of CGPK for each s of CGPK if(c和s有k-1条相同的分支 且concurrence( 且 令NewSet=NewSetU{ncgp} 并标识c和s为子模式; CGPK=NewSet; CGP=CGPUCGPK; While(NewSet is not null); (4) CGP即为并发间隙模式全集,删除其中的所有被标识为子模式的元素,得到最大集MCGP. (5) 在生成并发间隙模式的过程中,生成的并发间隙模式的数量会随着制定的mincon的递增而递减. 为验证PBcon算法的有效性和可行性,尝试在蛋白质序列上挖掘并发间隙模式.WildSpan算法首先挖掘出带有固定间隙的块集,块集的性质正好满足刚隙模式集的定义.因此,先用WildSpan挖掘出带有固定间隙约束的由蛋白质组成氨基酸为元素的块集(刚隙模式集)SetOfGP,进行基于蛋白质刚隙模式的并发序列模式挖掘,实现PBcon算法.需要指出的是,这里仅是使用WildSpan算法的中间结果作为进一步挖掘基础的刚隙模式集,而刚隙模式集的挖掘方法并不只限于使用WildSpan算法. 实验数据是从蛋白质功能位点数据库下载的蛋白质序列的文件.一条蛋白质序列是由A、 C、D、E、F、G、H、I、K、L、M、N、P、Q、R、S、T、V、W、Y 20个字符的字母表元素组成的线性序列,其中蛋白质序列是用PORSITE语言描述的fasta格式,一个文件包含多个fasta格式的蛋白质序列. 在CODEBLOCK软件上用C++实现了PBCon算法,并在内存为2G、CPU为2.40 GHz的Windows2007操作系统的环境上进行了蛋白质序列分析测试. 首先用WildSpan算法在数据源PS00627.fasta上生成包含99个刚隙模式的候选1-刚隙模式集,生成刚隙模式时minsup=48.86 %;然后用PBcon算法对其分别进行挖掘实验. 图1表示数据源PS00627.fasta用PBcon算法在不同并发度下并发间隙模式全集中的2、3、4、5分支并发的间隙模式个数.由图1可知:并发间隙模式集数随最小并发度mincon的增加而减少,表明并发度越大,所能挖掘的并发间隙模式的模式数量越少,基本呈递减趋势,与理论分析相符. 图1 数据源PS00627.fasta并发间隙模式变化曲线 图2是数据源PS00627.fasta用WildSpan算法挖掘W-模式的变化曲线.由图2可知:W-模式的挖掘结果随minsup的变化而变化,但是只在minsup=51.14 %时挖掘出一个W-模式. 图2 数据源PS00627.fasta W-模式变化曲线 图3是数据源PS00627.fasta用PBcon算法挖掘并发间隙模式集所需要的运行时间曲线.该曲线表明:随着并发度的增加,挖掘所需要的时间减少.这与随并发度的增加而产生的并发间隙模式集数减少有关. 图3 数据源PS00627.fasta运行时间变化曲线 表1是数据源PS00627.fasta用WildSpan算法和PBcon算法生成的W-模式和并发间隙模式集的对比. 表1 PBcon算法和WildSpan结果对比 在对PS00627.fasta的挖掘结果中,WildSpan仅在minsup=51.14 %时挖掘出1个W-模式P-x(3)-G-L-x(1,3)-S-S-A-x(146,267)-G-A-G;在PBcon算法挖掘中,当mincon=55.00 %时,挖掘出P-x(3)-G-L(137,142)S-S-A(144,146)G-x(2)-S-S(218,222)G-L-x-S(325,328)G-A-G(332,334),上述W-模式被包含其中.除此以外,PBcon算法在mincon=90.00 %时挖掘出1个2-分枝的并发间隙模式和一个3-分枝的并发间隙模式,说明它们之间的并发度很高,值得生物学家分析其中的可能信息.PBcon算法在mincon=80.00 %~50.00 %时均能挖掘出更多的多分枝并发间隙模式. 并发性是研究序列结构关系的重要特性.本研究将并发序列模式挖掘和间隙模式应用于蛋白质基序的结构关系挖掘.相比较WildSpan算法,PBcon算法可以找到更多的有意义的并发间隙模式.这有助于在蛋白质序列的组成中找到更多隐藏在结构之间的关系,可将其应用在预测蛋白质功能点的组合分析中.同时,PBcon算法与现有算法相比效率更高.今后将致力于找到更加优化的算法,提高效率,把并发序列模式应用到更多的类似蛋白质序列的序列分析数据中.2 PBcon算法分析
3 蛋白质序列并发间隙模式挖掘实验
3.1 数据准备
3.2 实验过程与分析


4 结 语
猜你喜欢
小资CHIC!ELEGANCE(2022年2期)2022-01-11
航空发动机(2020年3期)2020-07-24
家庭影院技术(2019年8期)2019-08-27
传媒评论(2018年5期)2018-07-09
计算机与生活(2018年3期)2018-03-12
延河(2017年7期)2017-07-19
阳光(2017年7期)2017-07-18
成长·读写月刊(2017年4期)2017-05-16
中国科技期刊研究(2017年2期)2017-05-14
浙江大学学报(工学版)(2015年2期)2015-05-30
