
以肝功能轻度异常为首发表现的高苯丙氨酸血症1例
2019-07-17 06:23:14曹丽丽崔莹春王丽旻
传染病信息 2019年3期
曹丽丽,崔莹春,王丽旻,董 漪,张 敏
高苯丙氨酸血症(hyperphenylalaninemia, HPA)是由于苯丙氨酸羟化酶(phenylalanine hydroxylase,PAH)或辅酶四氢生物蝶呤(tetrahydrobiopterin,BH4)缺乏导致血浆内苯丙氨酸(phenylalanine,Phe)浓度升高的一组最常见的氨基酸代谢性疾病,属于常染色体隐性遗传病[1-2]。本病在我国发生率8.5/10万[3],属于新生儿出生缺陷常规筛查疾病,主要影响神经系统发育,导致患儿智力发育落后、小头畸形、抽搐等。本病早期无明显的临床症状,以肝功能异常为首发症状表现的更是罕见。现将本院收治的1例以肝功能轻度异常为首发表现的HPA患者诊疗情况报告如下。
1 病例报告
1.1 病史 患儿,男,14岁,因“肝功能异常8个月”于2018年8月17日就诊于中国人民解放军总医院第五医学中心。2018年1月患儿在当地查体发现肝功能异常(ALT约为200 U/L,AST不详),给予保肝、降酶治疗,1月后复查肝功能正常并停药,此后患儿每月复查肝功能基本正常,直到2018年7月转氨酶再次升高,入住青海省妇女儿童医院查肝功能:ALT 111 U/L,AST 69 U/L,铜蓝蛋白17.6 mg/dl,血氨61.4 μmol/L。自身抗体、巨细胞病毒、EBV、肺炎支原体、柯萨奇病毒、单纯疱疹病毒、弓形虫、风疹病毒抗体、嗜肝病毒学指标均阴性;腹部及心脏彩超未见异常;胸片示肺纹理粗;串联质谱血、尿液筛查结果显示Phe增高。给予保肝、降酶等治疗,8月4日查ALT 81 U/L,AST 30 U/L。我院门诊以“肝损害原因待查”收入院。既往体健,家族中无遗传病及传染病史。
1.2 入院查体 体温36.7 ℃,脉搏76次/分,呼吸18次/分,血压111/73 mmHg(1 mmHg=0.133 kPa),身高169 cm,体质量47 kg,身体质量指数16.63 kg/m2,发育正常,营养良好,心、肺、腹查体未见阳性体征。初步诊断:肝损害原因待查,考虑先天性遗传代谢性疾病可能性大。
1.3 实验室检查及其他辅助检查 血常规:WBC 5.86×109/L、中性粒细胞百分比 55.24%、PLT 190.00×109/L、RBC 4.59×1012/L、HGB 122.00 g/L、中性粒细胞计数 3.23×109/L。尿、便常规检查未见异常。肝功能:ALT 80 U/L、AST 31 U/L、ALB 40 g/L、TBIL 7.4 μmol/L、DBIL 2.6 μmol/L、胆碱酯酶 8821 U/L、总胆汁酸17.7 μmol/L、ALP 308 U/L、γ-谷氨酰转肽酶23 U/L、AFP 2.57 ng/ml。肾功能、血脂、胆固醇、电解质、血糖、免疫球蛋白、蛋白电泳、血沉正常。贫血三项:叶酸 2.41 ng/ml、铁蛋白14.25 ng/ml,维生素B12正常。血氨 12.2 μmol/L、乳酸 2.72 mmol/L、铜兰蛋白 0.20 g/L、α1-抗胰蛋白酶1.50 g/L、血清铜10.9 μmol/L。(血、尿)巨细胞病毒DNA<100 IU/ml,EBV DNA<100 IU/ml。尿铜22.2 μg/24 h。自身抗体、嗜肝病毒学指标阴性。腹部彩超提示肝回声增粗。肝脏硬度值为6.8 kPa,脂肪衰减190 db/m。头颅MRI显示右顶叶腔隙性脑梗塞/缺血灶(急性期),左颅中窝脂肪瘤不除外。肝组织病理结果提示轻度慢性肝损害,肝脏病变程度相当于G0-1S1,不排除非嗜肝病毒感染。串联质谱尿液筛查:Phe检出明显(无具体数值);串联质谱血液筛查:Phe 143 μmol/L(正常值120 μmol/L),Phe/酪氨酸(tyrosine,Tyr)3.4(正常值2.0)。
1.4 诊治分析 患儿因“肝损害原因待查”收入院,患儿病史及常规检查排除病毒性肝炎、自身免疫性肝炎、酒精性肝病、药物性肝损伤、肝豆状核变性、抗胰蛋白酶缺乏及血色病等疾病。当地医院提示抗EBV-CA-IgG阳性,我院检查EBV DNA<100 IU/ml,肝穿病理G0-1S1结果显示可能为非嗜肝病毒感染,但临床表现不甚相符。当地行串联质谱血、尿液筛查曾提示Phe升高,我院复查串联质谱血、尿液筛查亦提示Phe检出明显,可能存在Phe代谢异常疾病,但患儿14岁,生长发育及智力均正常,无明确神经系统症状,与HPA临床表现同样不符。追问病史,患儿曾于2018年1月“晕厥”1次;且本次入院头颅MRI提示右顶叶腔隙性脑梗塞/缺血灶(急性期),结合血、尿液筛查提示Phe升高,有脑梗塞灶,初步诊断为HPA。
为进一步明确病因,经家长同意对患儿行基因序列测序,结果显示:患儿在苯丙酮尿症(phenylketonuria, PKU)相关基因 PAH存在c.1238G>C(鸟嘌呤>胞嘧啶)和c.157C>A(胞嘧啶>腺嘌呤)两处杂合突变,导致氨基酸改变(见图1)。人类基因突变数据库显示,突变位点c.1238G>C为致病突变[4],是引起该病的主要原因,突变位点c.157C>A致病性尚不明确。此外,本例患儿突变位点c.157C>A与既往文献报道突变点的突变点位置一致[5],但突变类型不同[c.157C>T(胞嘧啶>胸腺嘧啶)]。家系验证结果显示,此双杂合突变分别来自其父母,为复合杂合突变,符合常染色体隐性遗传规律。且患儿血 Phe> 120 μmol/L,Phe/Tyr> 2.0,符合 HPA诊断标准,故该患儿可明确诊断为PAH缺乏症。
后经保肝、低Phe饮食等治疗,患儿肝功能恢复正常,病情好转出院。出院后嘱患儿继续低Phe饮食,对其进行跟踪随访,患儿血Phe浓度持续在120~360 μmol/L之间,病情稳定,可正常生活及学习。
2 讨 论
PAH缺乏症是由PAH基因突变导致血浆内Phe升高的遗传代谢性疾病。PAH基因位于染色体12 q23.2[6],全长约90 kb,有13个外显子和12个内含子,转录后的mRNA含1353个碱基,翻译成451个氨基酸的酶单体,4个单体聚合形成具有功能的PAH。PAH存在于肝脏内,如肝内PAH活性降低或丧失时,Phe无法转化为Tyr、多巴胺和黑色素等正常代谢物,在体内积聚,对脑或神经系统造成不同程度的损害。该疾病呈常染色体隐性遗传,98%~99%的PKU患者是由PAH基因突变所致。我国PKU的发病率约为1/11 144,PAH突变在普通人群的携带率约为1/35[7]。
HPA患儿出生时大多表现正常,未经治疗的患儿3~4个月后逐渐表现出智力、运动发育落后,头发由黑变黄,皮肤变白,全身和尿液有特殊的鼠尿臭味,并可伴有湿疹、惊厥发作等症状,并且随着年龄增长逐渐显现出智力低下、行为缺陷及心理疾病等[8]。HPA目前是国家规定的新生儿时期进行的筛查疾病,早发现、早诊治对后期治疗预后较好。
HPA诊治共识提出诊断标准[1]。采用串联质谱测定血Phe、Tyr及Phe/Tyr[9],进行HPA的诊断:血Phe浓度>120 μmol/L及Phe/Tyr>2.0诊断为HPA[10-12];如仅有血Phe轻度增高,Phe/Tyr正常,排除其他疾病后须定期复查;如仅有Phe/Tyr>2.0,血Phe正常,不支持HPA。我国HPA分类标准:血Phe≥1200 μmol/L,经典型PKU;血Phe 360~1200 μmol/L为轻度PKU;血Phe120~360 μmol/L为轻度HPA。
本例14岁男性少年,因发现肝功轻度异常入院寻找病因,串联质谱检测发现血浆内Phe浓度升高(143 μmol/L),Phe/Tyr升高(3.4),符合HPA诊断标准。此时应鉴别是一过性特殊代谢状态、PAH缺乏症、还是BH4缺乏症引起的HPA。BH4是PAH羟化过程中所需要的一种辅酶,如其缺乏亦会导致Phe不能正常代谢,使血中Phe浓度增高出现神经系统症状,10%~15%的HPA可以归因于BH4缺乏症[13]。但仅仅依靠Phe浓度无法鉴别PAH和BH4缺乏症,须行尿蝶呤谱分析及二氢生物蝶啶还原酶(dihydrobiopteridine, DHPR)活性测定或基因检查来鉴别诊断。因我院检测条件有限,无法行蝶呤谱和DHPR活性分析,经家长同意后,对该病例直接进行了基因检测。结果发现在PAH基因外显子区域两处杂合突变:c.1238G>C(鸟嘌呤>胞嘧啶)、C.157C>A(胞嘧啶>腺嘌呤)。其中c.1238G>C为明确的致病突变, c.157C>A与文献报道的致病突变位点一致,但突变类型不同,已知的致病突变是c.157C>T(胞嘧啶>胸腺嘧啶)[4-5]。但本例的c.157C>A突变导致了氨基酸改变p.R53C(精氨酸>半胱氨酸),属非常可疑致病。家系验证结果显示此双杂合突变分别来自于父母,为复合杂合突变,符合常染色体隐性遗传规律。
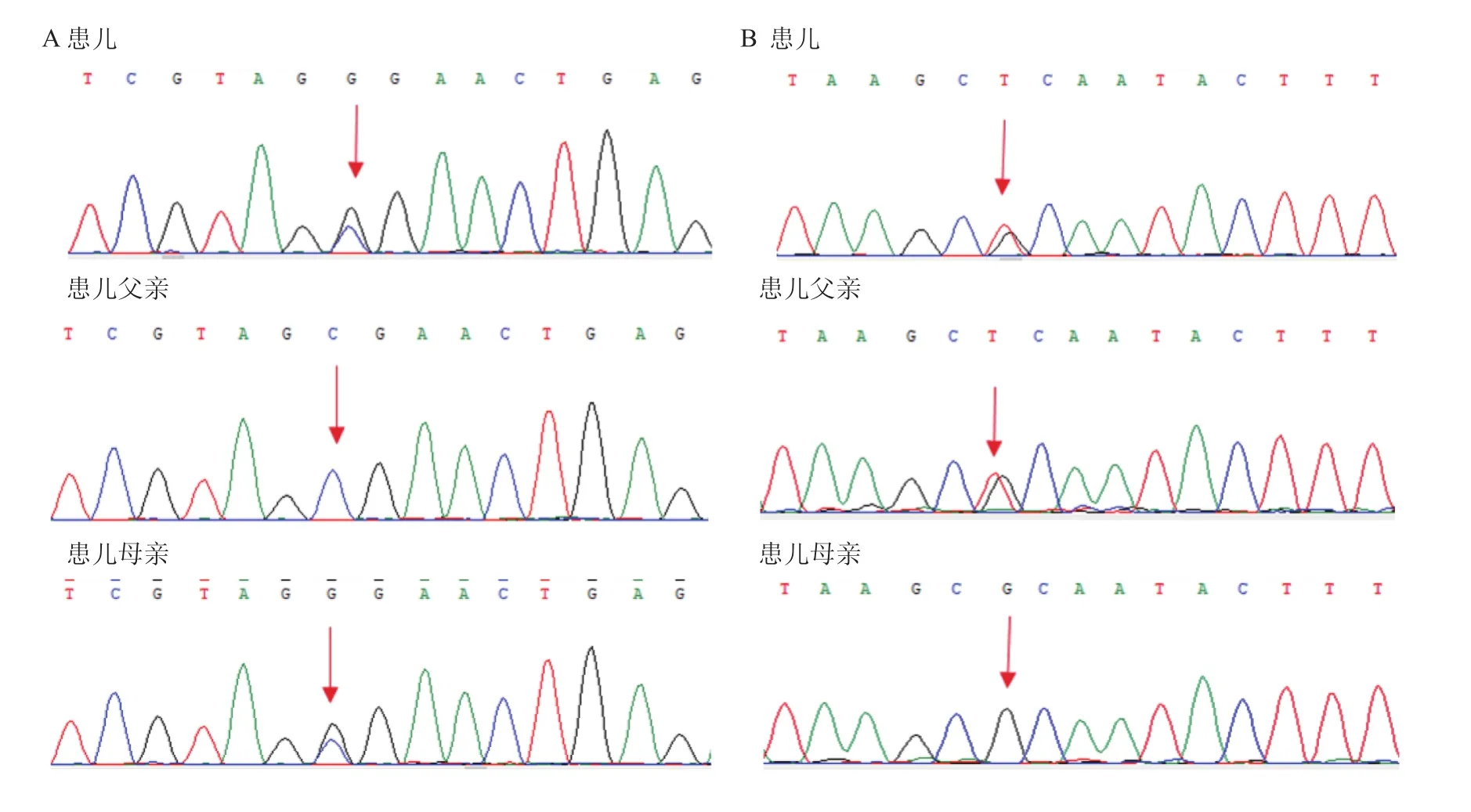
图1 PKU患儿及其父母PAH基因测序结果A.患儿及其母亲PAH基因可见c.1238G>C突变,患儿父亲PAH基因c.1238G>C未见突变;B.患儿及其父亲PAH基因可见c.157C>A突变,患儿母亲PAH基因c.157C>A未见突变Figure 1 Sequencing results of PAH gene in children with phenylketonuria and his parents
PAH目前已发现的基因突变数超过 950种(BIOPKU数据库)[14],这些突变包括错义突变、缺失突变、剪切突变等,大部分为错义突变,常导致蛋白错误折叠或催化功能损害,在种族和区域性也存在异质性,也有可能存在多态性[6]。R243Q是中国人以及朝鲜人最常见的突变位点[6],日本以 R423P发生率最高,R408W高发于东欧地区、不列颠群岛以及美国。IVS12+1G>A常见于丹麦和英国,斯堪的纳维亚(半岛)的高发位点为 Y414C,I65T常见于西欧。总之,世界范围内PAH突变差异巨大,表现出高度的遗传异质性。本患儿c.157C>A突变的基因位置与文献报道的致病位点一致,但核苷酸突变类型不同,估计这正是患儿病情轻、临床症状不典型的原因,可以推断c.157C>A突变虽然影响了PAH功能,但较c.157C>T突变对PAH功能的影响小。本患儿血Phe浓度轻度升高,随着年龄增长,其运动和智力均正常,并未对其造成明显的神经系统损害。其临床以肝功轻度异常为首发表现,按肝功异常原因待查诊断思路基本排除了病毒性肝炎、自身免疫性肝病、药物性肝损害等,肝穿病理很轻微,未发现特异性病变,考虑可能与肝脏内PAH缺乏导致肝细胞受损有关,但具体机制尚不明确。如果进行相应饮食治疗后肝功能复常,更能证明肝脏内PAH缺乏可能与肝功能异常相关,有待进一步随访。
典型的HPA临床症状主要以神经系统损害和智力发育落后为主,脑实质病变可能是脑皮质萎缩、脑白质脱髓鞘改变,MRI的T1加权图像上可显示脑室三角区周围脑组织条形或斑片状高信号区。本例曾出现“晕厥”,行头颅MRI提示右顶叶腔隙性脑梗塞/缺血灶(急性期),与常见PAH导致的病变不同,是否与本病直接有关,尚未见报道,有待密切监测、随访并进一步文献调研。
目前PAH缺乏症尚无治愈的方法,治疗原则是低Phe饮食。治疗指征各国不同,对血Phe浓度在360~600 μmol/L的未治疗者预后报道不同[15-16]。我国诊治共识建议血Phe浓度>360 μmol/L开始治疗。通过饮食控制Phe的摄入,减少血液及脑组织中Phe蓄积,避免不可逆的神经功能损害,治疗的年龄越小,预后越好,智能发育越可能接近正常人。经低Phe饮食治疗的患儿须密切监测血Phe浓度,使其浓度控制在相应年龄理想范围。除了监测血Phe浓度外,体格及智能发育评估也非常重要[17]。患者治疗期间的营养状况、心理、精神、行为、认知等方面的随访仍须要与多学科医生共同合作,预防营养缺乏及长期治疗产生的心理行为等障碍,提高生活质量。本患儿目前血Phe浓度143 μmol/L,生长发育良好,属于轻度HPA,告知低Phe饮食,建议其密切监测血Phe浓度,一旦高于360 μmol/L,须积极治疗。
总之,通过本例患儿肝功能异常待查的诊断,临床医生应了解PAH缺乏症亦有可能以肝功能异常为首发表现,须扩展诊断思路,以进一步提高临床医生对此类疾病不典型表现的辨识能力,达到早诊断、早治疗的目的。
猜你喜欢
今日农业(2021年16期)2021-11-26 06:05:34
吉林大学学报(理学版)(2021年3期)2021-05-26 02:24:00
种子(2021年3期)2021-04-12 01:42:22
家庭医学(下半月)(2019年9期)2019-10-12 08:03:58
猪业科学(2018年5期)2018-07-17 05:56:18
外语教学理论与实践(2016年1期)2016-06-11 05:51:48
百科知识(2015年13期)2015-09-10 07:22:44
质谱学报(2015年5期)2015-03-01 03:18:25
华东理工大学学报(自然科学版)(2014年1期)2014-02-27 13:48:29
影像科学与光化学(2014年1期)2014-02-23 05:51:38
