
HPLC法测定芪贞增免颗粒、蛋鸡宝中淫羊藿苷的含量
2019-07-16 01:41:12章安源章安雯张志民李有志
中国兽药杂志 2019年6期
章安源,章安雯,张志民,陈 玲,李有志∗
(1.山东省兽药质量检验所,山东省畜产品质量安全监测与风险评估重点实验室,济南250022;2.枣庄职业学院,山东枣庄277100)
淫羊藿是使用悠久的传统中药之一,其主要有效成分是黄酮类化合物淫羊藿苷[1]。现代药理学研究发现,淫羊藿苷具有多种生物活性[2],并有抗缺氧、抗氧化等多种生物学功效[3-4]。淫羊藿来源于小檗科淫羊藿属植物Epimedium L 干燥叶,以淫羊藿为主药的制剂很多,如芪贞增免颗粒和蛋鸡宝,相关标准仅用薄层和显微鉴别淫羊藿苷[5],淫羊藿药材种类多,鉴别困难。采收于夏秋的淫羊藿药材地上部分花果已脱落,仅存叶片和叶柄,药材中残存一些花序轴和果梗,可作为补充特征,若药材仅有叶片,可利用鉴别的特征就更少[6]。统计表明淫羊藿药材各药用部位中淫羊藿苷含量均为叶>根>茎[7],中成药在投料的过程中若投入淫羊藿茎及地下部分,仅靠当前的质量评价方法难以鉴别[8]。未对含量进行控制,不能全面反映制剂的质量水平,达不到制剂治疗效果。
本实验室首次应用HPLC 对芪贞增免颗粒和蛋鸡宝中淫羊藿苷进行定量研究,方法快速、准确。为控制本制剂的质量提供标准依据。
1 仪器与试药
1.1 仪器与设备 高效液相色谱仪Waters e2695(配二极管阵列检测器2998);AE-240 电子天平(瑞士Mettlrer Toledo 公司);KQ - 250DB 超声仪(昆山市超声仪有限公司);
1.2 试药与试剂 淫羊藿苷(批号:110737-201516,含量94.2%);购自中国食品药品检定研究院。甲醇、乙腈(Merck,色谱纯);水为超纯水。
2 方法与结果
2.1 色谱条件 色谱柱为Agilent Eclipso XDB-118 C18 柱(250 mm×4.6 mm,5 μm);流动相A:乙腈;流动相B:水[5];流速:1 mL/min;进样量10 μL;柱温为30 ℃。检测波长范围270 nm,结果见表1。

表1 梯度洗脱表Tab 1 Gradient elution
2.2 溶液制备
2.2.1 对照品溶液 精密称取淫羊藿对照品适量,加甲醇配制成浓度为100 μg/mL 的对照品溶液。结果见图2。
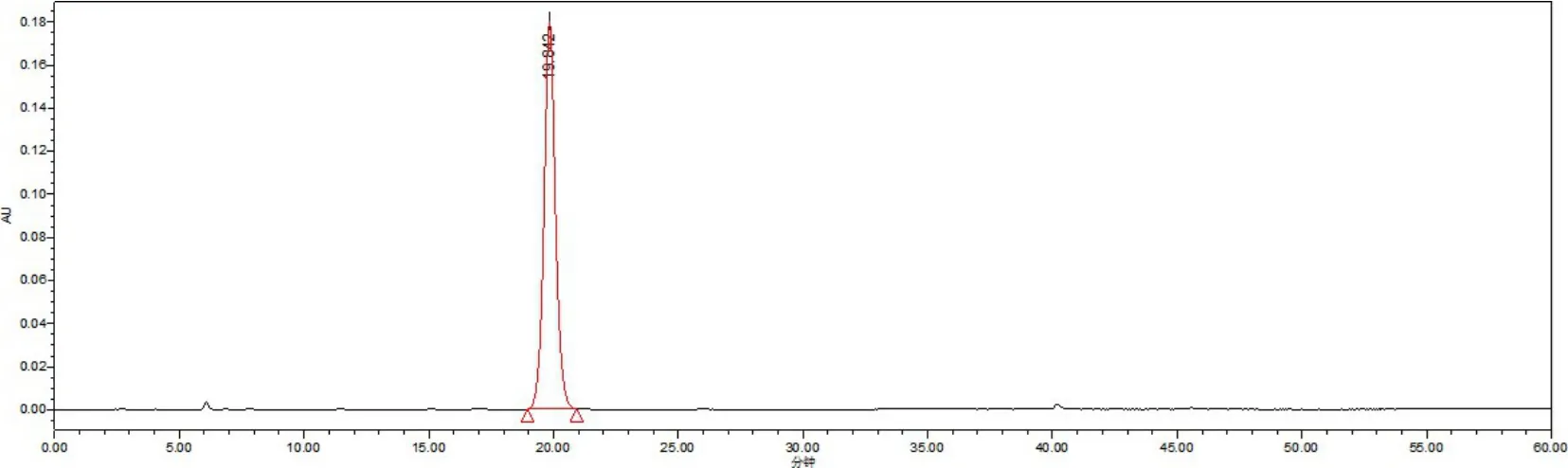
图2 淫羊藿对照品溶液Fig 2 Control solution
2.2.2 标准工作液的制备 取适量2.2.1 项下溶液,用初始流动相稀释,制备成系列浓度为5、10、25、50、100、200、250、400 μg/mL 标准工作液,绘制标准曲线。
2.2.3 供试品溶液制备 取样品充分研磨,精密称取1.0 g,置具塞锥形瓶中,精密加入40%甲醇25 mL,称定重量,超声(120 W,频率40 kHz),超声水浴温度40 ℃,处理30 min,放冷,用40%甲醇补足减失的重量,摇匀,滤过,取续滤液后再次精密加入40%甲醇25 mL,合并两次滤液后,0.22 μm 微孔滤膜滤过,即得[5]。对照品与供试品出峰时间相一致。结果见图3和图4。
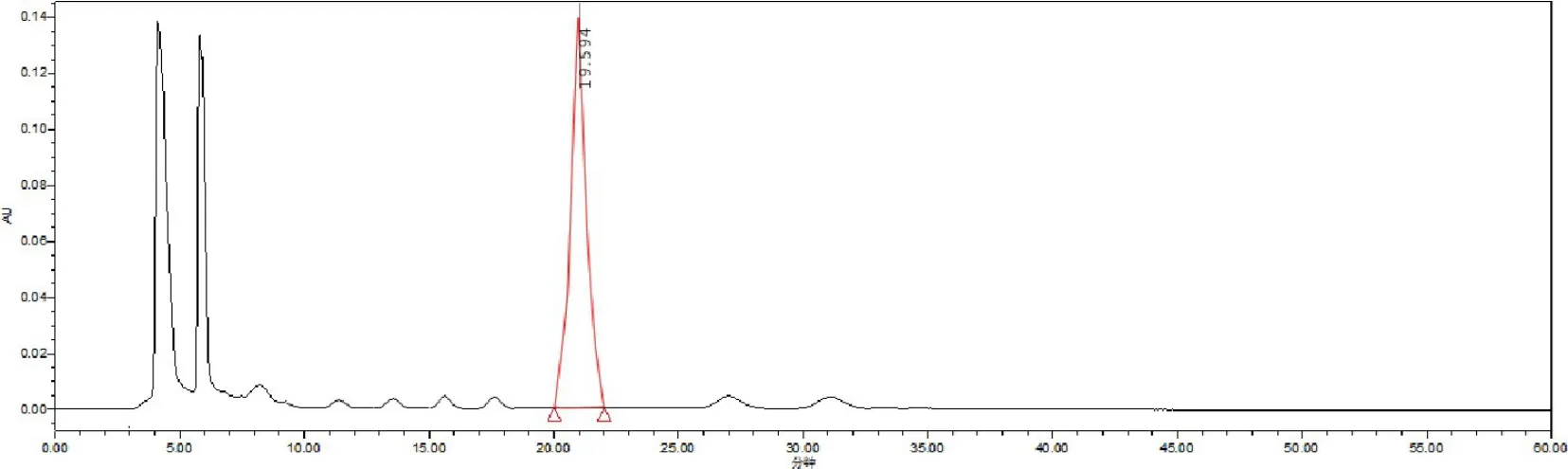
图3 芪贞増免颗粒Fig 3 Sample solution of Qizhenzengmian Granule

图4 蛋鸡宝Fig 4 Sample solution of Danjibao Powder
2.2.4 阴性样品 取除淫羊藿的其它药材适量,按样品的制备工艺和配方比例制备阴性样品,按2.2.3 项下方法制备阴性对照溶液,即得。
2.2.5 阳性添加样品 精密称取已知含量的同批样品(山东某兽药生物有限公司提供)6 份,分别精密加入适量对照品,置于25 mL 容量瓶中,按2.2.3项下方法混合均匀制成阳性添加样品溶液。
2.3 方法线性考察 分别精密吸取2.2.2 项下标准工作液10 μL,在2.1 项色谱条件下注入仪器,以峰面积为横坐标(X),浓度(μg)为纵坐标(Y),回归方程Y=0.0004X-0.2941,R2=0.9998;结果表明,淫羊藿苷在5~400 μg 与其对应的峰面积有良好的线性关系。
2.4 精密度试验 精密吸取对照品溶液10 μL 注入色谱仪,连续进样6 次。以峰面积计算,得出淫羊藿苷的RSD2.11%。表明仪器精密度良好,符合检测要求。
2.5 重复性试验 同批供试品(批号为20180201,山东某兽药生物科技有限公司提供),按2.2.3 项下制备6 份样品溶液,分别注入液相色谱仪,计算淫羊藿苷含量,RSD为1.4%,说明该方法重现性好,含量稳定。
2.6 稳定性试验 取2.2.3 项下同一供试品溶液(批号:20180201),室温下每隔4 h 连测6 次,进样体积20 μL,淫羊藿苷峰面积RSD分别为0.93%,0.89%、0.91%,表明本制剂在制备后的24 h 内成份较稳定。
2.7 回收率试验 取2.2.5 项下溶液2 mL,按2.1项下条件测定淫羊藿苷含量。回收率在92.02%~92.22%之间,变异系数在1.54%~1.88%之间,见表2-表3。表明该方法回收率较好,准确度和精密度可以满足检测需求。

表2 芪贞增免颗粒中淫羊藿苷回收率试验(n=6)Tab 2 Recovery of icariin of Qizhenzengmian granule
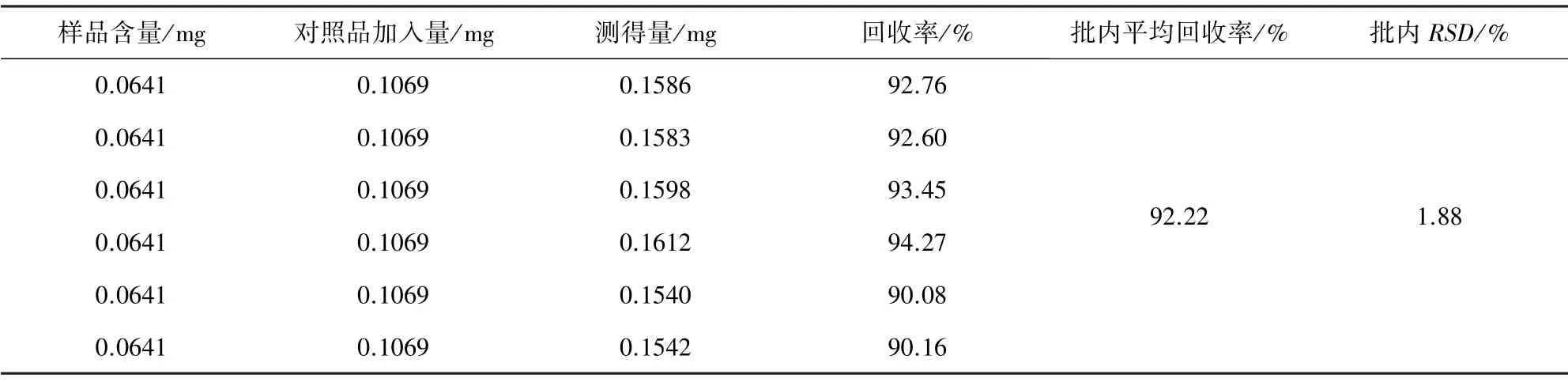
表3 蛋鸡宝中淫羊藿苷回收率试验(n=6)Tab 3 Recovery of icariin of Danjibao powder
2.8 样品测定 精密称取不同企业芪贞増免颗粒,蛋鸡宝样品,按2.2.3 项下方法制备供试品溶液,每份样品测定2 次,以2 次测定结果的平均值作为测定值,结果见表4。
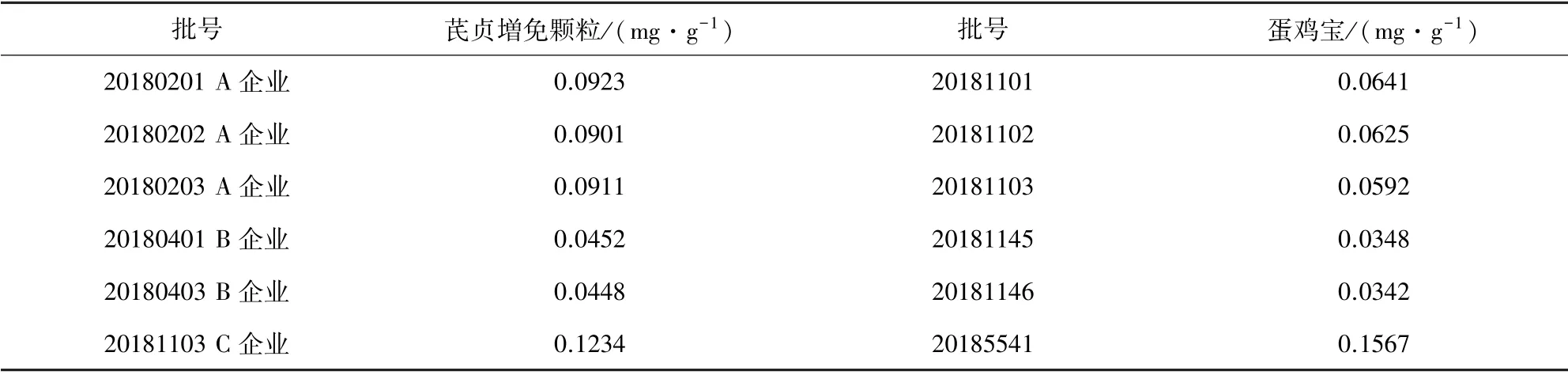
表4 样品测定结果(n=6)Tab 4 The resultof the sample content
2.9 检出限和定量限 取“2.2.4”项下溶液,色谱图显示无淫羊藿成分干扰,应用本试验建立的检测方法,添加对照品0.5 μg/mL,取与对照品图谱中相同保留时间的噪音信号平均值,信噪比(S/N)≥3,确定为方法的检出限。添加对照品10 μg/mL 显示信噪比(S/N)≥10,作为此方法定量限。
3 讨论与结论
3.1 供试品前处理提取方法的选择 Jang 等以提取溶液浓度、提取温度、溶液pH、液料比及提取时间为因素,发现在各因素中,提取溶液浓度及提取温度是影响提取率的主要因素[9]。试验中着重考察以上条件因素。选择6 种不同浓度提取溶剂:乙醇30%、40%、50%和30%、40%、50%甲醇[10-13],结果表明40%甲醇溶解样品不易结团,溶解性最好且提取率最高,检测样品杂质峰少。分别采用回流和超声不同方式提取,超声水浴温度控制在20 ℃、30 ℃、40 ℃,并考察了不同的提取时间[14-17]。回流提取1 h 后和超声提取30 min 淫羊藿苷即可提取完全,考虑到超声提取更加简便、快速、故超声处理30 min 来制备样品。分别提取样品2 次、3 次、4 次,超声频率40 kHz,水浴温度控制在40 ℃,试验结果显示提取2 次与3 次、4 次的淫羊藿苷含量差异无统计学意义,为节约时间,提取2 次样品即可满足实验需求[18]。
3.2 流动相的优选 《中国兽药典》2015 版二部中淫羊藿药材含量测定使用流动相是乙腈∶水(30 ∶70),但制剂中其它药材对其分离度有影响,杂质多,故比较以下5 种流动相组进行梯度洗脱:乙腈-水(25 ∶75)、乙腈-0.05%磷酸(27 ∶73)[15]、乙腈-水(50 ∶50)[16],乙腈-水(28 ∶72)[17]、乙腈-水(23 ∶77)[19]。经过比对发现使用乙腈-水(25 ∶75)梯度洗脱后淫羊藿苷峰分离度高,峰形对称,并无拖尾及其它杂质峰的干扰。使用其他流动相显示淫羊藿苷多在30 min 后出峰,出峰时间晚,此方法测定供试品方法稳定,准确度高,且节约时间。
3.2 色谱柱的选择 使用不同型号的十八烷基硅烷键合硅胶色谱柱进行比较,HypersilBDS-C18(250 mm×4.6 mm,5 μm)[15],Cosmosil C18(250 mm×4.6 mm,5 um)[18]、Agilent Eclipso XDB-118 C18 柱(250 mm×4.6 mm,5 μm)进行比较,后者普通C18柱即可满足淫羊藿苷测定方法的重复性需求,结果表明方法实用性及耐用性良好。
3.3 柱温的确定 25 ℃和30 ℃、35 ℃柱温进行考察,30 ℃柱温时,基线平稳,柱压稳定,对照品出峰时间与样品时间一致,选择30 ℃柱温符合检测要求。
3.4 影响淫羊藿苷含量测定的因素 淫羊藿苷含量测定结果表明来自于不同厂家芪贞增免颗粒和蛋鸡宝含量差别较大,分析可能是不同产地淫羊藿药材中淫羊藿苷的含量有差异[13],也可能与药材贮存有关[2],其次或于颗粒剂与散剂不同的工艺制备过程中对药物中有效成分有影响。
3.5 结论 采用现行标准中简单的显微鉴别特征和薄层色谱难以控制制剂和药效的稳定性,难确保临床疗效。应完善制剂中淫羊藿苷含量质量标准。本实验处理简捷,快速,方法可靠,重现性及回收率均符合要求,适合测定淫羊藿苷的含量,可有效控制芪贞增免颗粒、蛋鸡宝等方剂的内在质量。今后还需结合大生产继续积累数据,考察产品的稳定性。也有研究认为以多指标来评价淫羊藿质量更为科学[13,15]。
猜你喜欢
食品与发酵工业(2023年21期)2023-11-26 07:50:24
今日农业(2022年2期)2022-11-16 12:29:47
煤化工(2022年3期)2022-07-08 07:24:42
今日农业(2021年7期)2021-11-27 13:44:48
中成药(2018年12期)2018-12-29 12:26:00
中成药(2017年4期)2017-05-17 06:09:49
中国资源综合利用(2016年10期)2016-01-22 08:36:09
少儿科学周刊·少年版(2015年11期)2015-12-17 23:47:17
少儿科学周刊·儿童版(2015年11期)2015-12-17 03:53:46
郑州大学学报(理学版)(2014年2期)2014-03-01 04:20:54
