
气相色谱法同时测定海水中13种拟除虫菊酯类杀虫剂的残留量
2019-07-16 01:17:14张华威崔艳梅王倩韩典峰罗晶晶黄会李佳蔚宫向红
中国渔业质量与标准 2019年3期
张华威,崔艳梅,王倩,韩典峰,罗晶晶,黄会,李佳蔚,宫向红*
(1. 山东省海洋资源与环境研究院,山东省海洋生态修复重点实验室,山东 烟台 264006;
2. 淄博德安环境检测有限公司,山东 淄博 255000)
拟除虫菊酯类杀虫剂是一类高效、低毒的广谱杀虫剂[1],目前全世界已有几十种拟除虫菊酯类杀虫剂成功被商业化[2],其在农业生产、水产养殖等过程中被广泛运用并发挥了重要作用[3]。随着近年来部分高毒农药逐渐被禁用[4],拟除虫菊酯类杀虫剂因广谱、低毒和高效等优良特性在中国得到迅速推广,尤其已在农业及林业领域大范围使用,且使用量不断增加[4]。拟除虫菊酯类杀虫剂可随雨水及生活废水等途径进入水环境[5],造成水体污染。此外,由于拟除虫菊酯类杀虫剂具有亲脂性,对水生生物具有较高的毒性且容易在水生生物中富集,通过食物链的传递,进而对近海的鱼虾贝蟹类等水产品造成危害[6],破坏生态系统[7],甚至对人类健康造成严重威胁。因此,对海水中拟除虫菊酯类杀虫剂残留量进行监测十分必要。
目前,常用的检测拟除虫菊酯类杀虫剂的方法主要有气相色谱法[8-9]、气相色谱-质谱法[10]、气相色谱-串联质谱法[11-13]、液相色谱法[14]、液相色谱-串联质谱法[15]、分光光度法(UV-VIS)[16]、薄层色谱法(TLC)[17]和酶联免疫法(ELISA)[18]等,每种方法均有其优势和不足。分光光度法、薄层色谱法及酶联免疫法虽然检测步骤相对简单快速,但其可能出现假阳性结果,并且无法准确定量,阳性样品必须需用其他方法再做进一步的确证。气相色谱法、气相色谱-质谱法、气相色谱-串联质谱法、液相色谱法和液相色谱-串联质谱法等方法可对菊酯类杀虫剂进行准确定量,其中气相色谱-质谱法、气相色谱-串联质谱法及液相色谱-串联质谱法在定性方面相比于气相色谱法更加准确且抗基质干扰能力更强,但是由于大部分常用菊酯农药中均含有卤素原子等使其具有电负性的物质,因此使用气相色谱仪的ECD检测器检测可获得更低的检测限,且气相色谱法相对于质谱法定量更加准确,重现性更好。液相色谱法的定量限相比于气相色谱法更高,即在测定低浓度样品时存在劣势。因此,气相色谱法在实际检测海水中13种拟除虫菊酯类杀虫剂时更具优势。
已有研究中,拟除虫菊酯类杀虫剂的检测方法大多针对于果蔬[19-21]、茶叶[22-24]、水产品[25-27]和沉积物[28-29]等,而对海水中的拟除虫菊酯类杀虫剂残留量检测的报道较少。在海水中拟除虫菊酯类农药的测定相关的标准方面,HJ 698—2014《水质 百菌清和溴氰菊酯的测定 气相色谱法》[30]规定了水中溴氰菊酯的气相色谱测定方法;HJ 753—2015《水质 百菌清及拟除虫菊酯类农药的测定 气相色谱-质谱法》[31]规定了水中8种拟除虫菊酯类农药的测定方法;SL 740—2016《水质 甲萘威、溴氰菊酯、微囊藻毒素-LR的测定 高效液相色谱法》[32]规定了测定水中溴氰菊酯的高效液相色谱法。这些方法多是针对水体中2~8种拟除虫菊酯类药物的同时检测,而实际海水中通常存在更为广泛的拟除虫菊酯类药物。为更准确地检测海水中拟除虫菊酯类药物残留量,更加客观地反映某一区域水体中该类药物的总体残留状况,本研究采用二氯甲烷作为提取溶剂,经Florisil固相萃取柱净化和气相色谱法检测,建立了海水中13种拟除虫菊酯类杀虫剂的快速、灵敏且准确的检测方法,以满足海水中农药残留分析的要求,为海水质量监测提供技术支持。
1 实验方法
1.1 仪器与试剂
主要仪器:配有电子捕获检测器(ECD)的Agilent 7890A气相色谱仪购自美国Agilent公司;高速控温离心机购自美国Sigma公司;N-EVAPTM112氮吹仪购自美国Qrganomation Associates公司;Milli-Q Gradient超纯水仪购自法国Millipore公司;KQ-600E超声波清洗器购自昆山市超声仪器有限公司;旋转蒸发仪购自德国IKA公司;分液漏斗振荡器购自上海爱朗仪器有限公司;玻璃砂芯抽滤装置购自四川蜀牛公司;0.45 μm混合纤维素滤膜购自上海兴亚净化材料厂;分液漏斗(1 L)购自四川蜀牛公司;玻璃砂芯层析柱(24 mm×400 mm)购自四川蜀牛公司;NH2固相萃取柱(3 mL/500 mg)购自CNW公司;HLB固相萃取柱(3 cc/60 mg)购自美国Waters公司;Florisil固相萃取柱(6 mL/1 g)、DB-5毛细管色谱柱、HP-5毛细管色谱柱和DB-17毛细管色谱柱(30.00 m×0.25 mm×0.25 μm)购自美国Agilent公司。
主要试剂:正己烷、二氯甲烷、环己烷和乙酸乙酯(色谱纯,德国Merck公司);丙酮(分析纯,国药集团)。13种拟除虫菊酯类杀虫剂(七氟菊酯、丙烯菊酯、联苯菊酯、甲氰菊酯、三氟氯氰菊酯、氟丙菊酯、氯菊酯、氟氯氰菊酯、氯氰菊酯、氟氰戊菊酯、氰戊菊酯、氟胺氰菊酯和溴氰菊酯)标准品(纯度均≥98%,德国Dr. Ehrenstorfer公司);实验用水为超纯水。
标准储备溶液:称取适量拟除虫菊酯类杀虫剂标准品,用正己烷溶解,配置成1 000.0 μg/mL标准储备溶液,使用前用正己烷稀释至所需质量浓度,4 ℃保存,有效期为6个月。
混合标准储备溶液:准确移取13种拟除虫菊酯类杀虫剂标准储备溶液各0.5 mL,于50 mL容量瓶中,用正己烷稀释并定容至刻度,配制成13种拟除虫菊酯类杀虫剂均为10.0 μg/mL的混合标准储备溶液,有效期为2周。使用前用正己烷稀释至所需质量浓度。
1.2 样品提取与净化
1.2.1 样品处理
将待测海水样品用玻璃砂芯抽滤装置经0.45 μm混合纤维素滤膜过滤。
1.2.2 提取方法
准确量取500 mL过滤后的海水样品置于1 L分液漏斗中,加入30 mL二氯甲烷,振荡10 s并排气,置于分液漏斗振荡器上振荡300 s,静置分层,将下层有机相收集于150 mL三角瓶中,于上层水相加入30 mL二氯甲烷,按照同上步骤重复提取一次,合并下层有机相,待用。
1.2.3 净化方法
将上述提取液转移到填充无水硫酸钠的砂芯层析柱(无水硫酸钠柱为8 cm,干法填充)中,使其缓慢通过无水硫酸钠柱,收集于100 mL鸡心瓶中,再用10 mL二氯甲烷淋洗2次,合并淋洗液于100 mL鸡心瓶中,置于旋转蒸发仪于35 ℃浓缩至近干,用5 mL正己烷溶解残渣,待用。
取Florisil固相萃取小柱,依次用5 mL正己烷-丙酮-乙酸乙酯混合溶液(85∶5∶10,V/V)和5 mL正己烷活化,加入上述复溶液,5 mL正己烷淋洗,弃去全部流出液,用10 mL正己烷-丙酮-乙酸乙酯混合溶液(85∶5∶10,V/V)洗脱。洗脱液于40 ℃氮气吹至近干,用正己烷定容至1 mL,待气相色谱(GC)分析。
1.3 色谱条件
色谱柱选用DB-5(30.00 m×0.25 mm×0.25 μm)或性能相当者;载气为高纯氮气;流速1.0 mL/min;进样口温度230 ℃;检测器温度310 ℃;不分流进样;进样量1 μL。
升温程序:70 ℃保持1 min,以30 ℃/min升至210 ℃,以2 ℃/min升至233 ℃,以1 ℃/min升至250 ℃,保持10 min。
1.4 残留量的计算
按式(1)计算试样中13种拟除虫菊酯类杀虫剂残留量。
(1)
式中:X为海水中被测组分的残留量(ng/L);Ci为海水样品经提取净化浓缩后的待上机溶液中被测组分的测定浓度(ng/mL);V1为海水样品经提取净化浓缩后的待上机溶液的体积(mL);V为待测海水的体积(mL)。
2 结果与讨论
2.1 GC参数的优化
2.1.1 色谱柱的确定
大部分拟除虫菊酯类杀虫剂具有电负性,因此,其在带有电子捕获检测器(ECD)的气相色谱仪上具有更好的选择性和更高的响应值。通过对DB-5(30.00 m×0.25 mm×0.25 μm)、HP-5(30.00 m×0.25 mm×0.25 μm)和DB-17(30.00 m×0.25 mm×0.25 μm)毛细管色谱柱进行对比,发现DB-5毛细管色谱柱对13种拟除虫菊酯类杀虫剂的分离效果更好,因此本研究选用DB-5(30.00 m×0.25 mm×0.25 μm)作为检测色谱柱。
2.1.2 程序升温的确定
由于本方法所检测的物质种类达13种,且其中7种物质具有同分异构体,因此,需要对程序升温过程进行优化,避免出峰时间重合。实验结果表明,七氟菊酯、丙烯菊酯、联苯菊酯、甲氰菊酯、三氟氯氰菊酯和氟丙菊酯在DB-5毛细管色谱柱的出峰时间靠前;氯菊酯、氟氯氰菊酯、氯氰菊酯、氟氰戊菊酯、氰戊菊酯、氟胺氰菊酯和溴氰菊酯等均具有同分异构体,出峰时间靠后。因此,升温程序设定时前期快速升温,以快速排出大部分杂质,避免杂峰影响待测物质;中期缓慢升温,使联苯菊酯与甲氰菊酯、三氟氯氰菊酯与氟丙菊酯完全分离;后期缓慢升温,使各同分异构体分离。考虑到分析时间过久容易降低总体检测效率,最终本研究确定升温程序:70 ℃保持1 min;30 ℃/min升至210 ℃;2 ℃/min升至233 ℃;1 ℃/min升至250 ℃,保持10 min。
2.1.3 其他色谱条件的确定
载气流速可以较为明显地影响待测物质的出峰时间,较低的载气流速可使各待测物更易分离,然而过低的流速会延长各待测物的出峰时间,增加程序升温的时间将降低检测的效率;而较高的载气流速可以使待测物质的出峰时间提前,减少分析时间,提升检测效率。本研究中,检测物质种类较多,且半数具有同分异构体,较高的载气流速容易造成多数同分异构体的峰无法分离,影响检测准确性,因此,需要综合考虑载气流速与程序升温,最终本研究确定载气流速为1.0 mL/min。
一般情况下,进样口汽化室的温度最好高于待测组分的沸点,以保证待测组分均能汽化进入色谱柱;检测器温度应高于进样口及柱温箱的最高温度,保证通过色谱柱进入检测器的待测组分及杂质均能被去除,保护检测器不被污染。因此,本研究将进样口温度设置为230 ℃,检测器温度设置为310 ℃。
13种拟除虫菊酯类杀虫剂标准溶液(50.0 μg/L)的气相色谱图见图1。
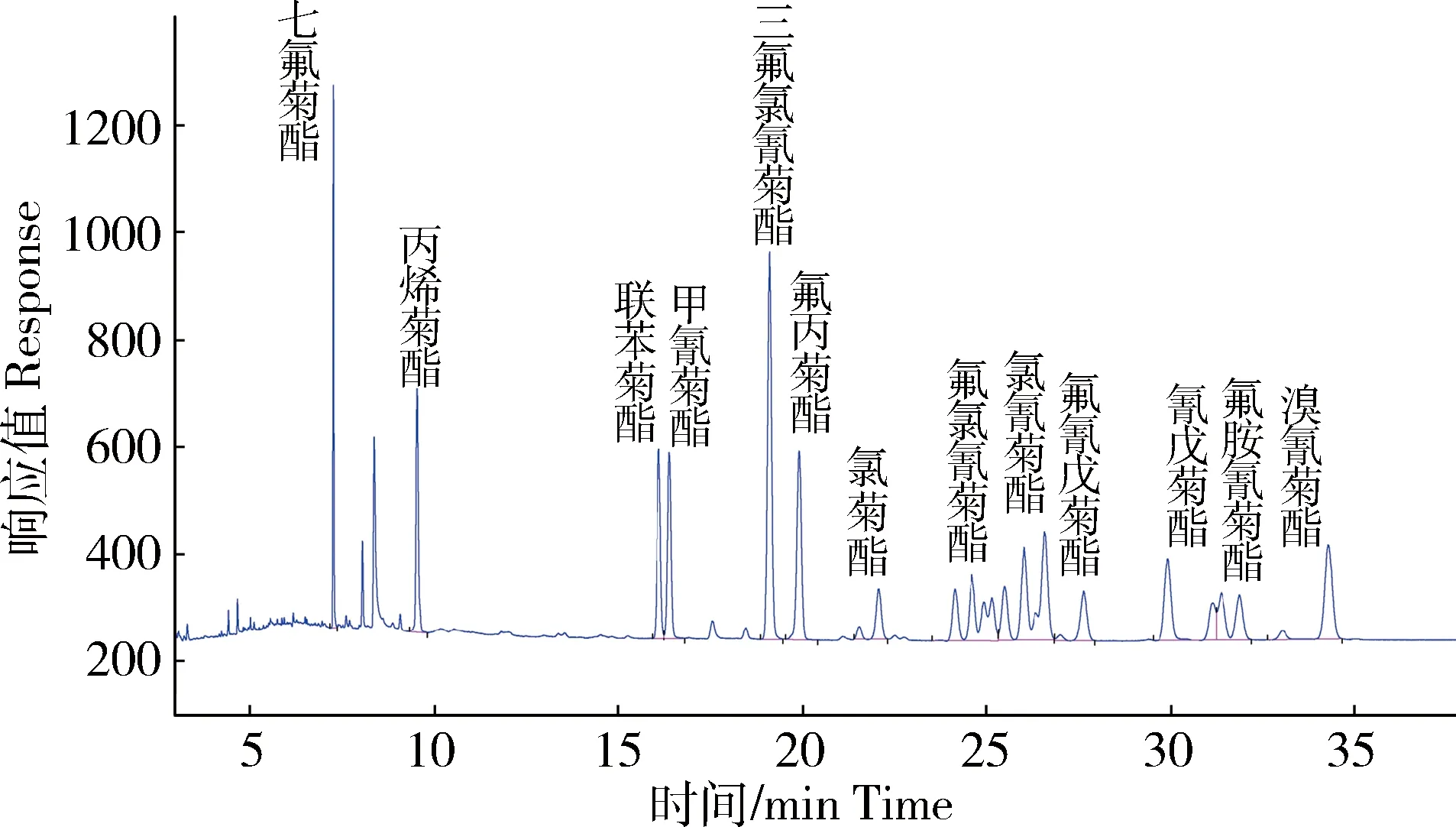
图1 13种拟除虫菊酯类杀虫剂标准溶液(50.0 μg·L-1)色谱图Fig.1 Chromatograms of 13 kinds of pyrethroid insecticides (50.0 μg·L-1)
2.2 样品前处理条件的优化
2.2.1 提取条件的确定
以空白海水样品进行加标回收试验,分别以二氯甲烷、正己烷和环己烷等3种有机溶剂为提取液,比较了不同提取溶剂对13种拟除虫菊酯类杀虫剂提取效率的影响(图2)。可以看出,二氯甲烷对13种待测物的提取效率均大于70%,优于正己烷和环己烷提取效果,因此选用二氯甲烷作为提取溶剂。
2.2.2 提取次数的确定
以二氯甲烷作为提取溶剂,分别用30 mL提取溶剂提取1次、30 mL提取溶剂提取2次、30 mL提取溶剂提取3次进行实验(图3)。结果表明,提取2次后,再增加提取次数回收率并没有明显增加。为提高检测工作效率,实验采用2次提取。
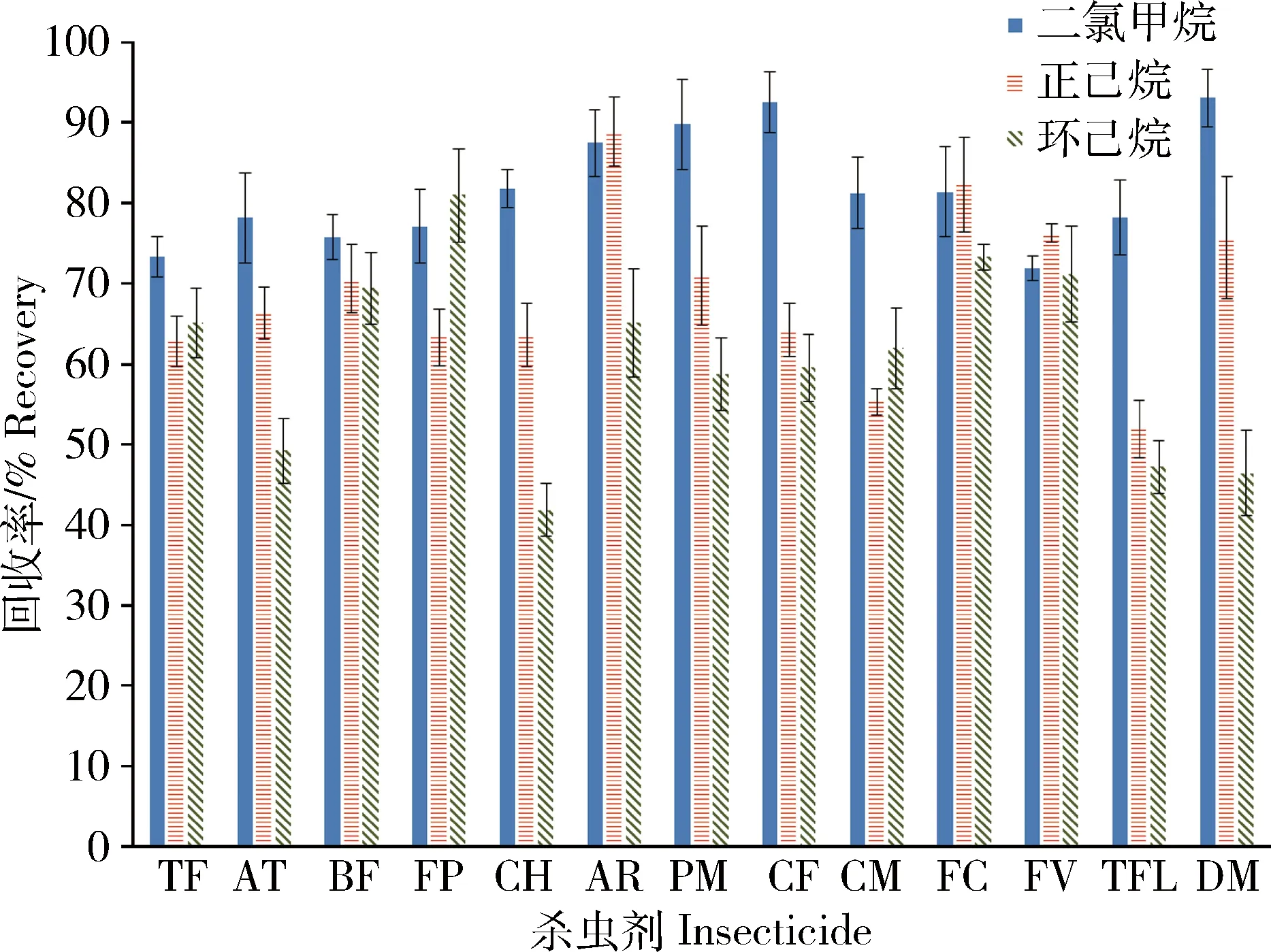
图2 3种提取溶剂对13种拟除虫菊酯类杀虫剂平均回收率的影响(n=3)TF:七氟菊酯; AT:丙烯菊酯; BF:联苯菊酯; FP:甲氰菊酯; CH:三氟氯氰菊酯; AR:氟丙菊酯; PM:氯菊酯; CF:氟氯氰菊酯; CM:氯氰菊酯; FC:氟氰戊菊酯; FV:氰戊菊酯; TFL:氟胺氰菊酯; DM:溴氰菊酯。下同。Fig.2 Effect of 3 kinds of extraction solvents on mean recoveries of 13 kinds of pyrethroid insecticides (n=3)TF: tefluthrin; AT: allethrin; BF: bifenthrin; FP: fenpropathrin; CH: cyhalothrin; AR: acrinathrin; PM: permethrin; CF: cyfluthrin; CM: cypermethrin; FC: flucythrinate; FV: fenvalerate; TFL: Tau-fluvalinate; DM: deltamethrin. The same below.
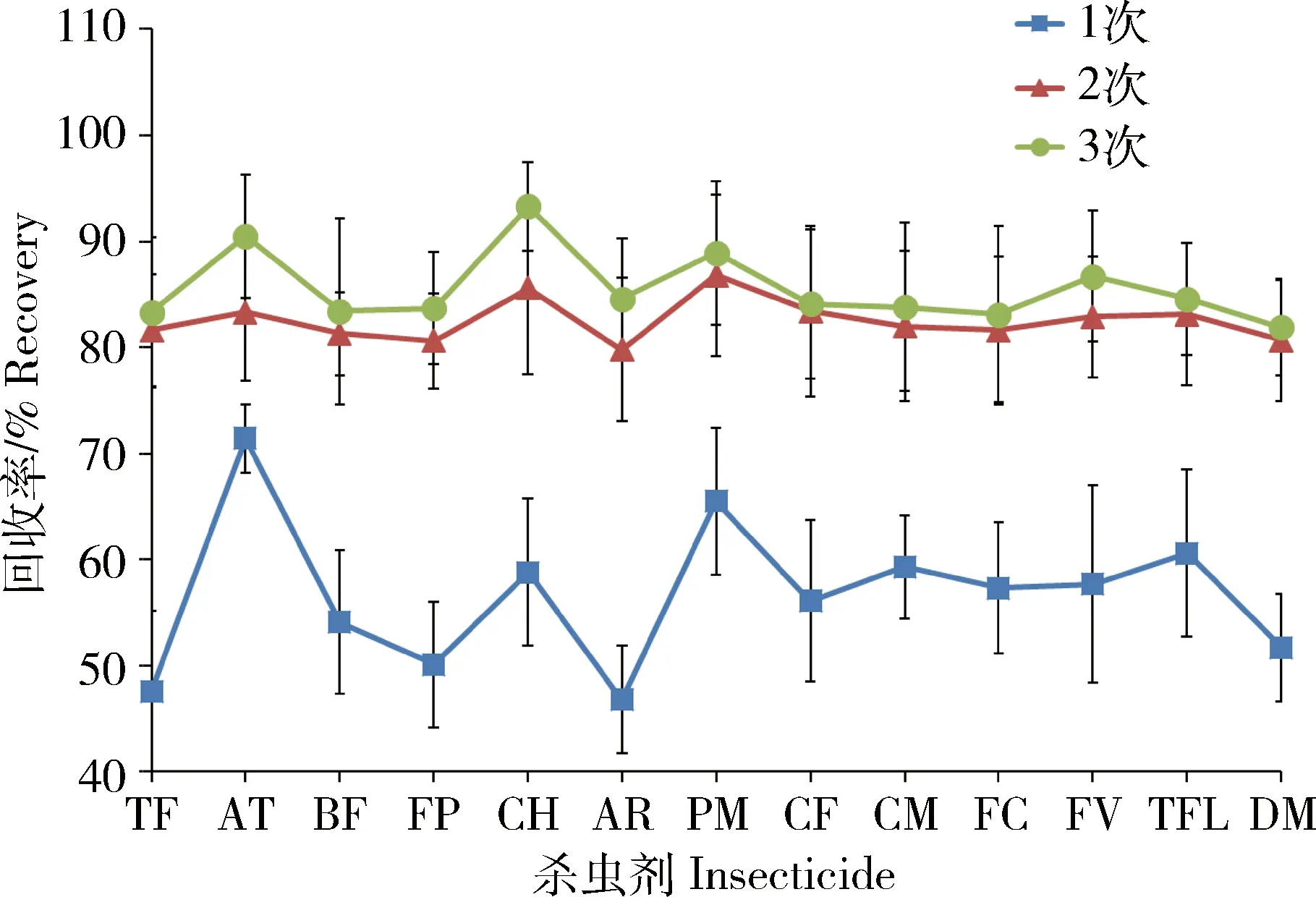
图3 不同提取次数对13种拟除虫菊酯类杀虫剂平均回收率的影响(n=3)Fig.3 Effects of different extraction times on mean recoveries of 13 kinds of pyrethroid insecticides(n=3)
2.2.3 净化方法的选择及优化
由于提取溶剂二氯甲烷呈中等极性,其在提取过程中会提取出大量杂质,可能污染色谱分析系统并对结果造成影响,因此需对提取液进行充分净化。本研究考察了Florisil固相萃取柱、NH2固相萃取柱和HLB固相萃取柱对提取液的净化效果(图4),结果表明,使用Florisil固相萃取柱净化样品时,13种拟除虫菊酯类杀虫剂的回收率均大于70%,且净化效果良好,优于NH2固相萃取柱及HLB固相萃取柱。因此,本研究选择Florisil固相萃取柱作为净化柱。
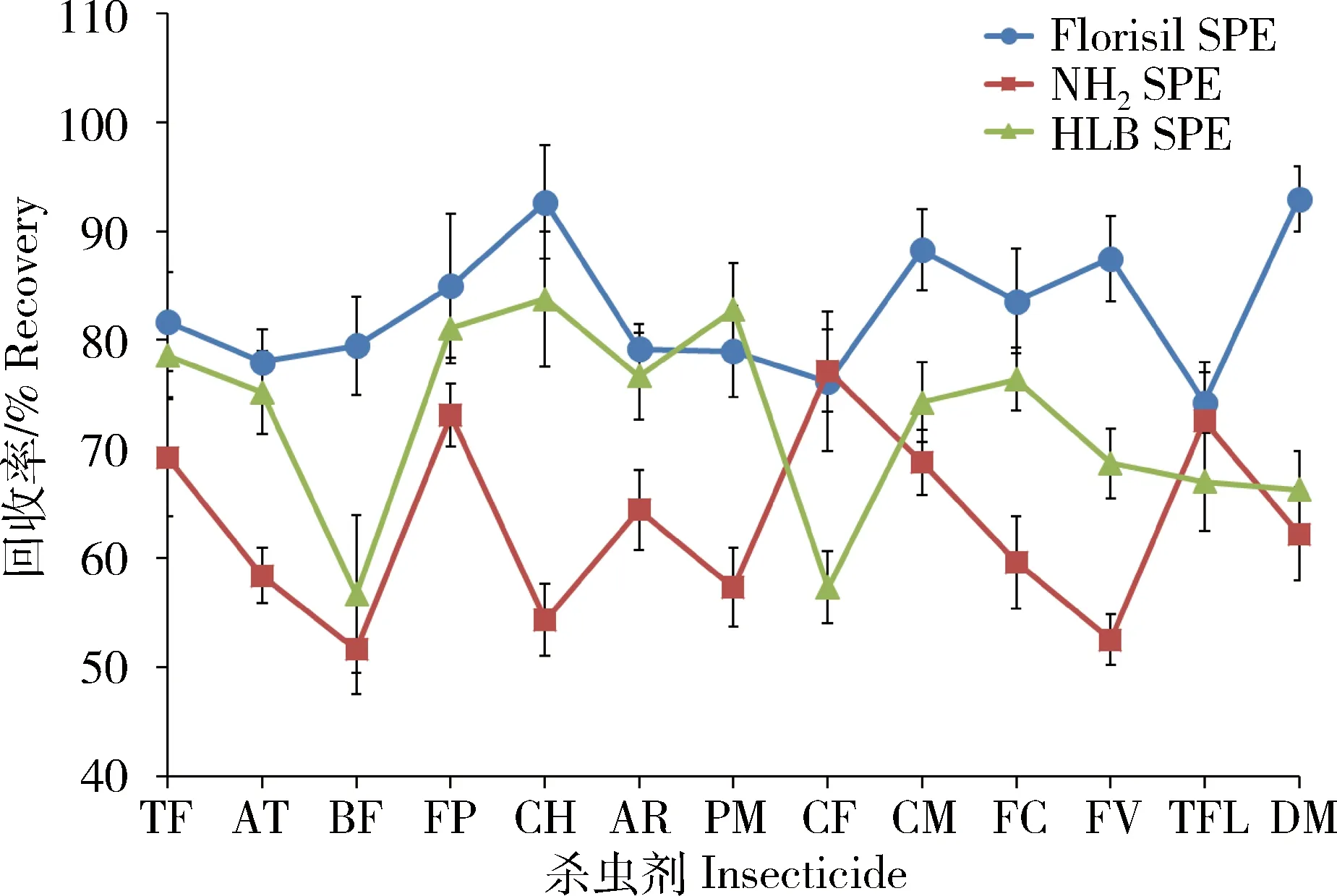
图4 3种固相萃取柱对13种拟除虫菊酯类杀虫剂平均回收率的影响(n=3)Fig.4 Effects of 3 kinds of SPE on mean recoveries of 13 kinds of pyrethroid insecticides (n=3)
根据拟除虫菊酯类杀虫剂呈弱极性的特征,分别选用正己烷-丙酮混合溶液(95∶5,V/V)、正己烷-乙酸乙酯混合溶液(90∶10,V/V)及正己烷-丙酮-乙酸乙酯混合溶液(85∶5∶10,V/V)作为Florisil固相萃取柱的洗脱溶剂进行试验。实验表明,使用正己烷-丙酮-乙酸乙酯混合溶液(85∶5∶10,V/V)作为洗脱溶剂时,洗脱效果最好,其回收率明显高于其他2种洗脱剂,同时实验发现10 mL正己烷-丙酮-乙酸乙酯混合溶液(85∶5∶10,V/V)即可使13种拟除虫菊酯类杀虫剂的回收率均达到70%以上。因此,本研究选择正己烷-丙酮-乙酸乙酯混合溶液(85∶5∶10,V/V)作为Florisil固相萃取柱的洗脱溶剂。
2.3 线性范围和灵敏度
取适量拟除虫菊酯类杀虫剂混合标准溶液,配制成2.5、5.0、10.0、20.0、50.0和100.0 μg/L系列浓度梯度的标准溶液,在1.3条件下依次测定,以各组分响应值为纵坐标、质量浓度为横坐标,进行线性回归分析。结果表明,13种拟除虫菊酯类杀虫剂在2.5~100.0 μg/L范围内线性良好。采用加标回收法确定各组分的检出限(LOD)及定量限(LOQ),以信噪比S/N≥3确定各组分LOD,以信噪比S/N≥10来确定各组分LOQ。本方法对13种拟除虫菊酯类杀虫剂的检出限均为5.0 ng/L,定量限均为10.0 ng/L(表1)。本研究所建立的方法灵敏度较高,检出限与定量限较HJ 698—2014、HJ753—2015及SL 740—2016等标准中相关的待测组分更低,在海水监测中能够提供更加准确的结果,适用于海水中拟除虫菊酯类杀虫剂的定量分析。
2.4 准确度和精密度
选取空白海水样品为测试基质,分别添加一定量拟除虫菊酯类杀虫剂混合标准溶液,使其质量浓度分别为10.0、50.0和100.0 ng/L,按本研究的方法进行测定,每个水平做6个平行样品,并计算加标回收率,结果见表2。结果表明,13种拟除虫菊酯类杀虫剂在海水中的加标回收率为76.6 %~107.1%,相对标准偏差为3.9%~12.7%。海水空白样品与加标回收样品(10.0 ng/L)气相色谱图分别示于图5和图6,由图可见,实验条件下,待测组分能与杂质有效分离,满足海水中13种拟除虫菊酯类杀虫剂的分析要求。

表1 13种拟除虫菊酯类杀虫剂的线性方程、线性范围和相关系数Tab.1 Linear equations, linearity range and correlation coefficient of 13 kinds of pyrethroid insecticides
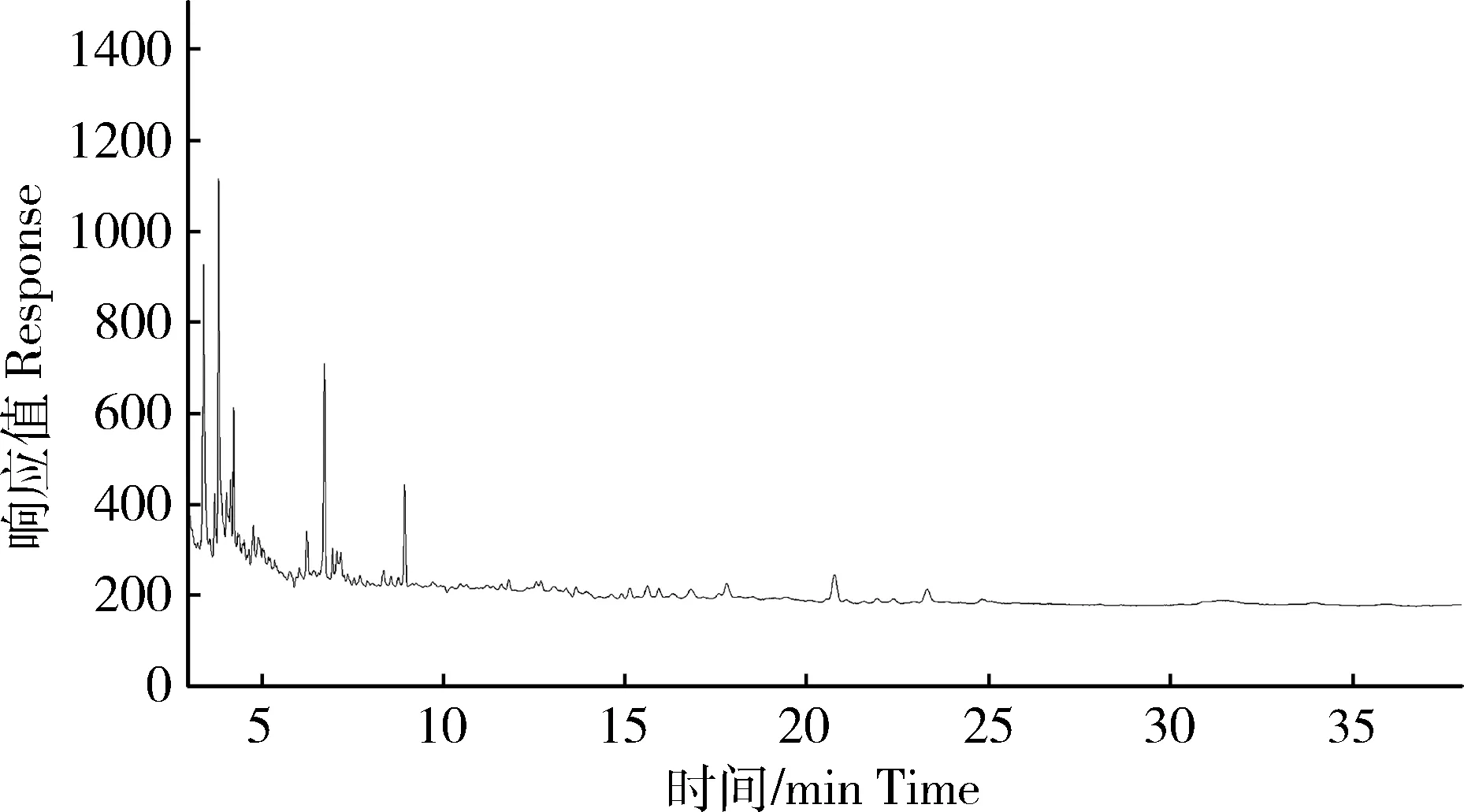
图5 空白海水样品色谱图Fig.5 Chromatograms of blank samples in seawater
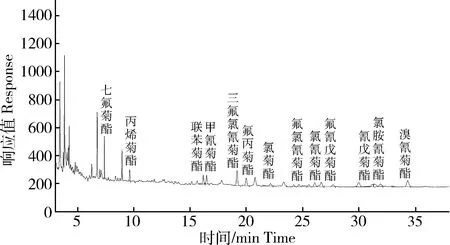
图6 海水中13种拟除虫菊酯类杀虫剂加标样品(10.0 ng·L-1)色谱图Fig.6 Chromatograms of spiked samples with 13 kinds of pyrethroid insecticides in seawater (10.0 ng·L-1)
2.5 实际样品测定
使用本方法对山东省某沿海地区采集的海水样品进行测定,其中部分样品检出甲氰菊酯、氯菊酯、氯氰菊酯及溴氰菊酯等(图7)残留,选取其中3个样品,实际测定结果见表3。
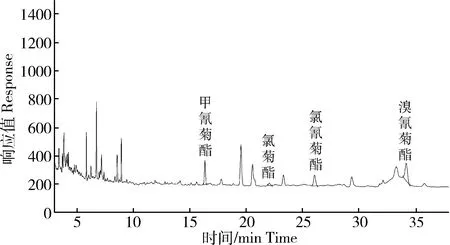
图7 海水样品的色谱图Fig.7 Chromatograms of seawater samples
3 结论
本研究建立了可同时检测海水中13种拟除虫菊酯类杀虫剂残留量的方法。方法采用二氯甲烷作为提取溶剂,Florisil固相萃取柱净化,外标法定量,13种拟除虫菊酯类杀虫剂的检出限均为5.0 ng/L,定量限均为10.0 ng/L。本方法与目前常用方法相较,可同时检测更多拟除虫菊酯类杀虫剂种类(13种),且被测组分的定量限更低,在对未知样品的实际分析中有望得到更全面的分析结果。
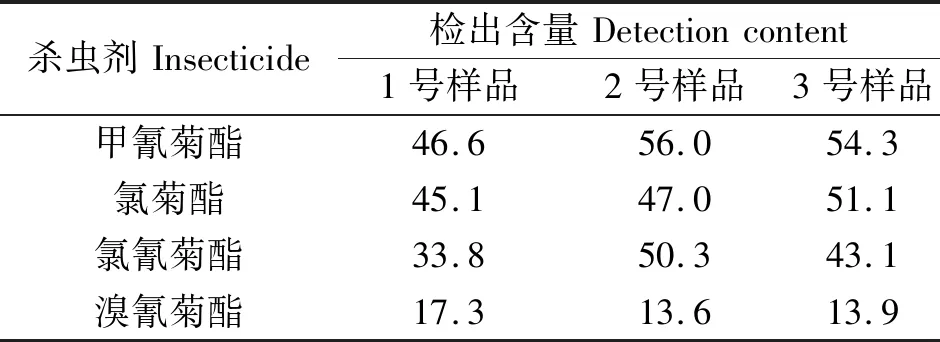
表3 海水样品中拟除虫菊酯类杀虫剂测定结果Tab.3 Contents of pyrethroid insecticides in seawater ng·L-1
猜你喜欢
江西农业学报(2022年8期)2022-11-04 07:37:02
乡村科技(2022年2期)2022-03-25 14:56:16
中国油脂(2020年3期)2020-04-10 02:08:54
中国医药指南(2017年3期)2017-11-13 02:59:17
上海农业学报(2017年3期)2017-04-10 12:39:20
无机化学学报(2016年8期)2016-12-06 09:05:14
科学种养(2016年4期)2016-04-19 03:47:43
化学分析计量(2016年1期)2016-03-14 00:35:19
分析测试学报(2015年3期)2016-01-13 06:18:20
食品科学(2013年8期)2013-03-11 18:21:28
