
基于Katz增强归纳型矩阵补全的基因-疾病关联关系预测*
2019-07-11 07:28浦建宇
计算机与生活 2019年7期
浦建宇,陈 蕾,2,3+,邵 楷
1.南京邮电大学 计算机学院,南京 210023
2.江苏省无线传感网高技术研究重点实验室,南京 210023
3.南京航空航天大学 计算机科学与技术学院,南京 210016
1 引言
疾病的发生与遗传、生活环境等很多因素有关,其中很大一部分疾病的产生与特定的基因有着密不可分的关系。生活中常见的癌症[1-3]、老年痴呆症[4]、糖尿病[5]等都属于多种基因缺陷导致的疾病。因此,发现疾病的致病基因,对于了解疾病发生原因、疾病的临床诊断和早期的预防治疗有着重要作用,也是人类基因组研究的重要目标,具有极大的科学与社会意义。另一方面,在当前的生物医药领域,率先发现疾病的相关致病基因,对于抢先研发出疾病的治疗手段和治疗药物有着不可忽视的作用,其所带来的经济效益也是巨大的。
早期开展基因-疾病关联研究都是基于临床及生物实验的方法进行的,这种方法通常会耗费大量的人力物力,不仅极大地限制了致病基因研究的发展,也严重影响着相关公共数据集的数据质量。例如,广泛使用的人类孟德尔遗传数据库(online Mendelian inheritance in man,OMIM)[6]和遗传关联数据库(genetic association database)[7]都只记录了极少部分基因-疾病间已经确定存在的关联关系,绝大部分基因-疾病间是否存在关联关系并未可知,从而一方面导致数据集中基因-疾病间的已知关联数据极为稀疏,另一方面导致数据集存在严重的数据偏斜问题,也就是说这些数据集只包含部分基因-疾病间确定的有关联关系(本文称之为Positive关系),并没有包含任何基因-疾病间确定的无关联关系(本文称之为Negative关系)。对那些未知的基因-疾病关联关系(本文称之为Unlabeled关系),需要预测它们之间是否存在关联。这类问题在机器学习领域通常称之为PU(positive and unlabeled)学习问题,现有研究已经表明负类(Negative)关系的缺失将严重影响着PU学习问题的学习效果[8]。而近年来,通过高通量测序、生物医学文本挖掘等手段,可以获得大量如基因阵列信息、基因内在特性、基因间相似性信息、疾病间相似性信息等有用的生物信息。此类信息的出现,也为研究新的预测方法来缓解以上不足提供了契机。首先,开发出了Katz这种基于网络相似度度量的方法,其通过融入基因间相似性信息、疾病间相似性信息和基因-疾病关联信息构建基因-疾病异构网络,通过在异构网络上预测来缓解数据稀疏的缺陷。但该方法对于未连接到网络中的节点无法进行有效预测并且会受到所构建的网络质量的影响[9-10]。之后,Natarajan等人[11]转而引入机器学习领域流行的归纳式矩阵补全(inductive matrix completion,IMC)方法来对基因-疾病关联关系进行预测,该方法利用基因和疾病的特征信息,能够有效克服冷启动问题。然而,该方法不仅遭受着数据稀疏的影响,而且也受到PU问题的影响。
针对以上问题,本文提出了一种基于Katz增强归纳型矩阵补全的基因-疾病关联关系预测(Katz boosted inductive matrix completion for gene-disease associations prediction,KIMC)模型。该模型的动机是利用传统的Katz方法来优化新近提出的归纳式矩阵补全方法,本质上属于步进式基因-疾病预测范型,包括基于Katz方法的预估计和基于归纳式矩阵补全方法的精化估计两个步骤。具体地,首先利用Katz方法基于所构建的基因-疾病异构网络对所有基因-疾病对进行Unlabeled关系的关联预估计。由于所估算出的关联评分数据中接近于1的数据可视为正关联信息,接近于0的数据可视为负关联信息,因此通过Katz预估计不仅缓解了数据稀疏缺陷,也缓解了PU问题对后续归纳式矩阵补全方法的影响。然而,受限于所构建的基因-疾病异构网络质量,基于Katz方法预估计出的基因-疾病关联信息不可避免地包含了一定程度的噪声。为了克服这些噪声对归纳式矩阵补全方法的影响,本文将弹性网正则化技术[12]引入新近提出的归纳式矩阵补全方法以增强其鲁棒性,进而利用改进的弹性网正则化归纳型矩阵补全模型来精化基因-疾病关联预测效果。OMIM数据集上的实验表明,本文提出的KIMC方法与其他几种竞争性方法比较,不仅在查全率和查准率上有显著提高,同时也能解决基因-疾病关联预测中常见的冷启动问题。
本文的主要贡献如下:
(1)提出了一种基于Katz增强归纳型矩阵补全的基因-疾病关联预测模型。该模型不仅融合了Katz方法和归纳型矩阵补全方法的优点,而且通过引入弹性网正则化机制增强了模型的容噪性能,能有效缓解传统方法易遭受的数据稀疏和PU问题的影响。
(2)采用近邻前向后向分裂技术设计了一种有效的弹性网正则化归纳式矩阵补全优化算法,同时从理论上证明了该算法的收敛性。
(3)OMIM数据集上的多组实验结果表明,所提出的KIMC模型不仅能够取得比现有预测方法更好的预测效果,而且能解决针对新疾病或新基因进行有效预测的冷启动问题。
2 相关工作
在过去的十几年间,已经提出了许多基于不同基因-疾病数据集的致病基因预测算法。主要分为基于网络相似度度量的方法和基于机器学习的方法。
Wu等人[13]提出了CIPHER(correlating protein interaction network and phenotype network to predict disease genes)方法,其假设在相互作用网络中更接近的两个基因可能会导致更相似的疾病。可以用基因相似性来解释疾病相似性,利用整个疾病相似性网络和PPI(protein-protein interaction)网络计算得到一个得分,通过这个得分衡量一个基因是特定疾病致病基因的可能性。Li等人[14]对随机游走方法进行改进,提出基于异构网络的随机游走(random walk with restart on heterogeneous network,RWRH)模型。首先利用基因间相似性信息、疾病间相似性信息和基因-疾病关联信息构建基因-疾病异构网络,这个方法充分考虑到了整个网络的全局信息。用一个随机游走粒子沿着网络连接关系进行扩散来捕获节点间的相似性,从而计算基因和疾病间的关系。基于Li等构建的基因-疾病异构网络,Singh-Blom等人[15]引入在社交网络分析中广泛使用的Katz方法,在异构网络上利用两个节点间不同步长的游走路径数量来计算节点间的相似性,从而预测基因和疾病的关联关系。Wang等人[9]以及Zou等人[10]对上述基于网络相似度度量的方法进行了比较细致的分析比较,这些方法通过计算网络中的候选基因和疾病节点之间的相似度来预测基因-疾病关联。这类算法的优点是能够将不同类型的基因相似性信息和疾病相似性信息融入到基因-疾病异构网络中,增强数据信息量;其缺点也很明显,对于那些没有连接到异构网络中的基因和疾病节点,不能有效预测,同时依赖于构建高质量的生物网络模型。
基于以上方法的局限,一些研究者又提出了基于机器学习的方法。例如:Singh-Blom等人[15]提出了CATAPULT(combining data across species using positiveunlabeled learning techniques)方法。该方法是一种监督机器学习方法,通过训练带偏置的SVM(support vector machine)分类器,进行基因-表型关联分类,从而挖掘出致病基因。接着,Natarajan等人[11]提出IMC方法,能够从基因微阵列数据、基因功能相互作用数据、不同物种的同源基因-表型数据中提取基因特征;从疾病相似性网络,疾病的临床表现数据,大量的医学文献中获取疾病特征,融入该方法中,弥补标准矩阵补全(matrix completion,MC)只能依赖于现有的可观察到的关联关系进行预测的局限,使得该方法具有一定的归纳性,能够对新的基因和疾病进行预测,解决了MC方法会遇到的冷启动问题,相较于之前提出的方法,预测效果有了很大的提升。
3 预备知识
本章主要介绍现有的几种不同的基因-疾病关联预测方法。
本文的主要目标是预测疾病的潜在致病基因,现在所使用的基因和疾病数据集通常只有少量的已知基因-疾病关联。通常,构建一个已知基因-疾病关联矩阵P∈RNg×Nd,如下:

行和列分别对应基因和疾病,Ng表示基因总数,Nd表示疾病总数,Pij=1表示基因i与疾病j之间存在关联,Pij=0表示基因i与疾病j之间关联未知(可能存在关联,也可能不存在)。由于该矩阵包含大量未知关联,因此所构建的基因-疾病关联矩阵是一个极度稀疏的矩阵,同时由于这里只有正关联数据,因此该问题是一个典型的PU学习问题。主要的任务就是通过设计有效的方法,将其中的未知关联预测出来,以达到预测致病基因的目的。
3.1 Katz方法
Katz方法类似于CIPHER[13]、RWRH[14]等算法,这些方法的本质都是基于网络相似性度量的算法。具体说来,Katz方法是基于基因和疾病关系网络计算基因和疾病间的相似性评分,并根据相似性评分对疾病对应的基因进行排序,选出合适的候选致病基因。Katz方法在社交网络关系预测中有着很成功的应用[16],它利用两个节点间不同步长的游走路径数量来计算节点间的相似性,在基因和疾病关系网络中,也是通过同样的方法计算节点间的相似性评分。
这里,利用基因-基因相似性网络、基因-疾病关联网络、疾病-疾病相似性网络构建一个基因-疾病关系异构网络,然后在异构网络上利用Katz方法预测基因-疾病关联。异构网络结构如图1。图示异构网络的邻接矩阵表示为:

其中,G表示基因-基因相似性网络;D表示疾病-疾病相似性网络;P表示基因-疾病关联网络。

Fig.1 Structure of heterogeneous networks图1 异构网络结构
由于在网络中基因Gi与疾病Dj之间有直接关联的数量并不多,因此,需要通过计算节点间不同长度路径的数量来表示基因与疾病之间的关联关系。(Cl)ij表示基因Gi到疾病Dj之间路径长度为l的路径数量。在C上定义节点间相似性如下:

其中,β为一个非负常数,用来控制不同长度路径的影响,β的取值范围为(0,min{1,1/‖‖C2})。将式(3)转换为矩阵形式,则相应的关联评分矩阵可表示如下:
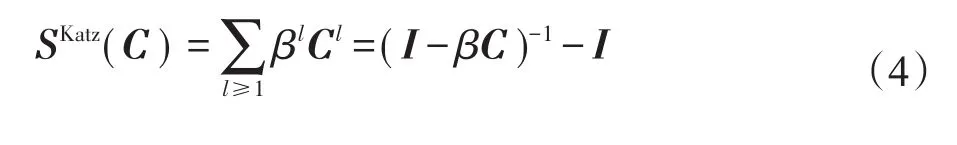
但是,在Katz方法中,没有必要去考虑所有长度的路径数量,因为较短路径长度的路径传达的节点之间的相似性信息更多,而距离较远的节点所传递的信息很少,所以只需要考虑有限路径长度的和。已有研究结果表明[17],较小的k值(通常取k=3或k=4)能够表现出很好的性能。在实验中,取k=3,取出对应的基因-疾病相似性Katz评分矩阵可表示为:

利用式(5)求基因和疾病间的评分。该方法将基因-基因相似性网络、疾病-疾病相似性网络这类辅助信息融入到基因-疾病异构网络中,有效提高了预测的效果。
3.2 标准矩阵补全(MC)
由于Katz等基于网络的关联预测方法的明显缺陷,提出利用矩阵补全理论进行基因-疾病关联预测。最初,利用MC方法预测基因-疾病关联关系,该方法将目标矩阵分解为两个低秩矩阵W∈RNg×k和H∈RNd×k的乘积,其中k≪Ng,Nd。因此,预测基因-疾病关联可以写成求解以下优化问题:

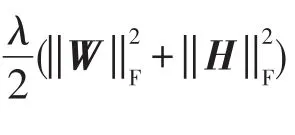
由于使用现有生物数据集构建的基因-疾病关联矩阵P是非常稀疏的。从OMIM数据库中获取的数据集中,大多疾病只有一个已知相关基因,大多数基因没有相关疾病。在这里,利用标准矩阵补全不能预测关联矩阵中那些完全没有元素的行和列,即遭遇冷启动问题。
3.3 归纳型矩阵补全(IMC)
由于使用标准矩阵补全预测基因-疾病关联时,所利用的数据类型单一(只利用已知基因-疾病关联),诸如生物医学文献,功能注释,蛋白质-蛋白质相互作用,不同物种的同源表型,基因微阵列等大量生物特征信息得不到有效利用。在预测时会遭遇冷启动问题,预测效果也不理想。针对以上问题,需要寻找一种能够有效利用这类基因和疾病的特征信息。Yu等人[18]提出的多标签学习问题能够很好地利用此类特征信息。在多标签学习中,需要学习一个低秩线性模型Z∈Rd×L,其中每一个样本(基因)由d个特征表示,并且有L个标签(疾病)。如果x∈Rd表示一个基因的特征向量,对应疾病j的预测可表示为xTZj,其中Zj表示矩阵Z的第j列。
将IMC[19]模型应用于基因-疾病关联预测问题,IMC假设通过将与其行和列实体相关的特征向量应用于低秩矩阵来生成关联矩阵,用P中观察到的元素来恢复Z。令分别表示基因i和疾病j的特征向量,表示Ng个基因的训练特征矩阵,其每一行代表一个基因的特征向量,表示Nd个疾病的特征训练矩阵,其每一行代表一个疾病的特征向量。IMC将建模为,需要恢复低秩矩阵Z,即Z=WHT,其中因此,基因-疾病关联预测建模为解决如下问题:

一个在训练数据中不存在的新疾病j′,如果有其特征向量yj′,那么对于所有基因i,能够计算出其所有的关联Pij′。同样,对于一个新的基因也是如此,而且能够有效解决MC方法所遭遇的冷启动问题。当特征数量很大时,取一个较小的k值,此时需要学习的参数个数小于fg×fd,在标准矩阵补全中,需要学习的参数个数为(Ng+Nd)×k,不难发现,IMC中需要学习的参数不依赖于基因和疾病的数量,仅取决于基因和疾病的特征数量。
MC问题可以看成是IMC问题的一个特例,即当基因的特征矩阵X为一个大小为Ng的单位矩阵,疾病的特征矩阵Y为一个大小为Nd的单位矩阵。在这里,使用交替最小化(即固定W求H或固定H求W,交替迭代求解)求解式(7),目标函数是一个凸函数,当W或H中的一个固定时,求解只有一个变量(W或H)的凸函数时,使用共轭梯度下降法求解。
3.4 基于Katz增强归纳型矩阵补全
由于现有的基因-疾病数据的极度稀疏性以及基因-疾病数据库中大多数只记录确定基因-疾病关联,现有的方法都会遭受数据稀疏和PU问题的影响。因此,需要寻求一种较为稳定的方法,能够缓解基因-疾病关联数据稀疏问题的影响以及PU问题的影响。
于是提出了集成Katz方法在基因-疾病异构网络上的关联预测和归纳型矩阵补全模型的KIMC方法。首先,在构建异构网络时,能够从被业界广泛认可的数据库中获取已被证实的基因-基因相似性信息和疾病-疾病相似性信息,与基因-疾病关联信息一起构成异构网络。相比使用特征信息的方法,Katz方法使用的这类信息能够更直接地传达基因-疾病相关信息。融合IMC方法,增强预测效果的同时,又不会失去其具有归纳性的特点,将问题建模为:


由于受构建的网络质量影响,引入残差矩阵R会带来一部分噪声,直接使用归纳型矩阵补全求解会影响预测效果和稳定性,因此引入矩阵弹性网正则化[12]来缓解这个问题,将求解残差R建模为:

进一步可将问题(10)转换成等价的罚函数形式:

本文拟采用近邻前向后向分裂(proximal forward backward splitting,PFBS)[20]技术对问题(11)进行优化求解。不妨令:

则问题(11)可形式化为如下一般形式:

根据PFBS规则,可对Z进行如下方式的迭代求解:

其中,δ为更新步长,且:

根据文献[21],对于矩阵B∈Rfg×fd和常数τ>0,有:

因此,对Z的迭代更新可转换为:

进一步,文献[20]的定理3.4表明:
命题1若最优化问题(13)的最小值存在且0<δ<2/Lf,则对任意初始参数Z0,解序列(14)收敛到式(13)的最小值。其中Lf为函数F2(Z)的Lipschitz连续梯度,即对于一个凸函数F(X∃),Lf>0,对∀X1,X2,有如下不等式成立:
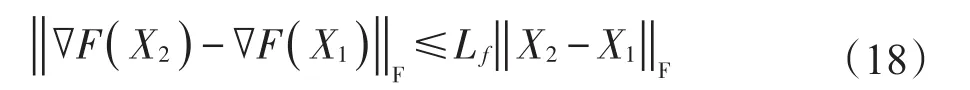
根据命题1,如果可以找到一个常数Lf>0并且使F2(Z)满足式(18),则解序列(14)收敛,则KIMC算法收敛,根据文献[22]引理1证明如下:
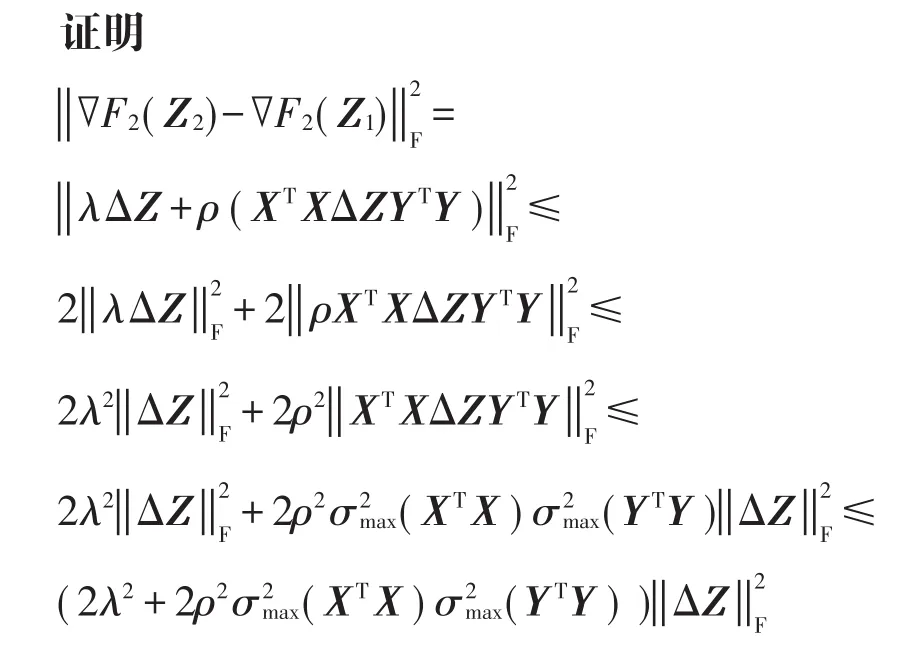
因此,Lipschitz常数为:

本文将无弹性网正则化项的KIMC模型和加入弹性网正则化项的KIMC模型分别表示为KIMC1和KIMC2,其中KIMC2求解过程如算法1所示。
算法1基于Katz增强归纳型矩阵补全
输入:基因和疾病特征矩阵X、Y,关联矩阵P,采样下标集合Ω,基因相似性矩阵G,疾病相似性矩阵D,参数β、δ、ρ、λ以及迭代次数Maxiter。

4 实验结果与分析
在本章中,介绍实验所用到的基因-疾病数据集和基因与疾病特征来源,以及基因-疾病关联预测所通用的评价标准,并对实验结果进行详细分析,比较几种方法的性能。
4.1 数据集与特征
本文使用的基因和疾病信息来自OMIM数据库,该数据库不仅收录了以孟德尔方式遗传的所有单基因病的相关资料,而且还收录了染色体病、多基因病、线粒体病方面的资料,涵盖病种丰富。其还提供了已知有关致病基因的连锁关系,染色体定位,结构与功能信息,同时描述了各种遗传病的临床信息,其信息更新及时,具有权威性。实验使用文献[15]所提供的基因-疾病数据集,该数据集包括从OMIM数据库采集的基因-疾病关联关系,其中有12 331个基因,3 209个疾病,共有3 954个已知基因-疾病关联;12 331个基因的基因-基因相似性信息和3 209个疾病的表型-表型相似性数据(即疾病-疾病相似性数据)。另外,本文所需的基因特征和疾病特征可以从不同类型、不同来源的生物数据中提取。例如,从基因微阵列数据,基因功能相互作用数据,不同物种的同源基因-表型数据中提取基因特征;从疾病相似性网络,疾病的临床表现数据,大量的医学文献分析数据中获取疾病特征。面对这类复杂的数据,通常会利用PCA(principal component analysis)进行降维来提取基因和疾病的主要特征,本实验使用文献[11]提供的基因和疾病特征。
4.2 评价指标与方法
与上文提到 Katz[15]、MC[11]、IMC[11]方法一样,实验使用3折交叉验证进行评估。评价预测性能时,使用top-r排序的方法(即对预测结果中每一个疾病列对应的基因评分值由大到小排序,取前r个基因作为对应疾病的候选致病基因)与另几种基因-疾病关联预测方法比较。在评价不同方法性能时,通过取不同阈值r所对应的疾病相关的致病基因,对比测试集中记录的已知关联,比较每种方法的查全率(Recall)。计算公式如下:

同时,也需要对实验结果的查准率(Precision)进行分析,其计算公式如下:

其中,TP表示测试集中已知基因-疾病关联中被正确判定的关联数量,FN表示测试集中已知基因-疾病关联中没有被正确判定的关联数量,FP表示未知基因-疾病关联中被判定为存在关联的数量。在现在的生物学研究领域中,希望在一个较小的阈值范围取得好的预测效果,通常取r≤100。其次,在评估预测方法全局性能同时,相较于一些被广泛研究的基因和疾病,研究者更关注那些存在研究空白的新基因和新疾病,希望能够不断发现有价值的新的基因和疾病关联,以促进医学研究的发展。因此,这里也关注各种方法对于新基因(即在数据集中仅有一个已知关联,但在训练时没有关联的基因)和新疾病(即在数据集中仅有一个已知关联,但在训练时没有关联的疾病)的预测能力。同时,为进一步验证本文所提出方法的有效性,选取8种常见病的前10个候选基因,与数据库及文献报道进行了对比分析。
4.3 全局性能
实验中对比了最近提出的几种基因-疾病关联预测方法:MC方法、IMC方法、Katz方法。3折交叉验证的查全率(Recall)结果如图2(a)所示,其中横坐标表示不同阈值r的取值,纵坐标表示查全率。本文提出的KIMC1方法和KIMC2方法在取不同的阈值r时性能都优于其他几种对比方法。在阈值取r=100时,几种方法的查全率分别为:MC方法为6.7%,Katz方法为11.3%,IMC方法为23.2%,KIMC1方法为26.5%,KIMC2方法为27.6%。加入弹性网正则化的KIMC2方法相较于之前提出的集成基因疾病特征的IMC方法有了一定提高。本文提出的方法同时集成了Katz方法和归纳性矩阵补全方法的优势,整体表现有了进一步提高,同时从图中可以看出加入弹性网正则化,可以有效缓解数据噪声的影响,提高了预测效果和稳定性。其次,这里同样给出了实验结果的查准率-查全率曲线图(precision-recall curves)。如图2(b),其横坐标为查全率,纵坐标为查准率。从图中可以发现,当查全率大于4%时,在相同的查准率下,KIMC1和KIMC2的查全率相较于其他3种方法都有一定提升。这里也对比了有/无弹性网正则化时不同阈值下的曲线,可以发现加入弹性网正则化后的KIMC2相较于KIMC1查准率也显著提高。

Fig.2 Overall performance at different thresholdr图2 取不同阈值r时的全局性能
4.4 新基因和新疾病预测
在基因-疾病关联关系预测中,往往会存在一个很容易被忽视的问题:现有的数据库中记录的多数都是认知度和关联度很高的基因和疾病,只有少部分关联单一的基因和疾病,因此在实验评估时,这类认知度和关联度较高的基因和疾病往往被预测出的几率更大,而在现实中研究者更关注那些处于研究空白的基因和疾病。因此,这里仅关注那些在数据集中已知关联单一的基因和疾病,在训练时将这些已知关联隐藏,以此来展现不同方法对新基因和新疾病的预测能力。在阈值r≤100的范围内,新基因查全率如图3(a)所示,其横坐标表示不同阈值,纵坐标表示新基因查全率。在阈值范围为0<r≤45时,Katz方法利用基因-基因相似性网络和疾病-疾病相似性网络作为辅助信息时,相较于IMC,预测效果更好。因为在异构网络中,此类数据能够更直接地反映出基因和疾病之间的关联。而IMC将不同的基因和疾病数据提取特征使用时,在此阈值范围内表现欠佳。当r>45时,IMC方法的预测效果明显提高,利用特征信息进行预测的优势得到体现。本文提出的KIMC1方法和KIMC2方法集成了Katz方法和IMC方法的优点,在提高预测效率的同时,使其在不同的阈值范围内预测表现更稳定。当r=100时,KIMC2方法的新基因查全率为17.4%。新疾病查全率如图3(b)所示,其横坐标表示阈值,纵坐标表示新疾病查全率。从图中可以发现,KIMC1方法和KIMC2对于新疾病的预测能力也优于其他几种对比方法。
4.5 一些常见病的Top-10致病基因预测

Fig.3 Recall at different thresholdrfor new genes and diseases图3 取不同阈值r时的新基因和新疾病的查全率
上述对新基因的预测能力的分析仅在OMIM数据库中的已知基因-疾病关联数据集上进行验证,对于一些没有记录在数据库中的致病基因无法进行评估验证,因此整体效果会偏低,同时对基因间的关联性也无法进行分析。这里选取几种现实生活中常见疾病排名前10的致病基因预测结果进行分析,对本文提出方法的效果进一步补充说明。这里选取了8种常见病,分别为:白血病(leukemia)、阿尔兹海默病(alzheimer disease)、抗胰岛素症(insulin resistance)、前列腺癌(prostate cancer)、精神分裂症(schizophrenia)、乳腺癌(breast cancer)、胃癌(gastric cancer)、结肠癌(colorectal cancer)。实验时,将训练数据中这8种疾病的相关致病基因信息全部隐藏(即这8种常见病对应的列全部置“0”),预测的疾病前10个候选致病基因如表1所示,表中疾病后的数字(如MIM:601626)表示其在OMIM数据库中对应编号,基因后的数字(如PAX6(5080))表示基因在NCBI数据库中对应编号。表中基因顺序按照预测评分由大到小排列。通过对表中候选致病基因的分析,可发现该方法预测出的致病基因并不仅限于基因-疾病关联数据集中已记录的基因,还能预测出一些后期研究发现的疾病相关基因。如与阿尔兹海默病相关的基因有PSEN1、PSEN2[4]等,这些已被证实的疾病相关基因在表中加粗显示。其次,从表中可以发现,这8种疾病的前10预测基因之间有很高的重叠度,有些基因在8种疾病中相互共享,如TP53、KRAS、RAD51已被证实与多种癌症的产生有密切关系[33],因此有充分理由相信,这些共享的基因实际上反映了不同疾病间的病因关联。即这类共享的基因会导致多种疾病的发生。通过对这类共享基因的分析,进一步验证了KIMC方法的预测结果能够展现出一些基因的共性。因此,KIMC方法能够为研究人员发现致病基因和研究致病基因间的关联提供有效的参考。
5 总结

Table 1 Prediction of top-10 candidate pathogenic genes for KIMC表1 KIMC预测前10个候选致病基因
本文提出了一种基于Katz增强归纳型矩阵补全(KIMC)模型的基因-疾病关联预测算法。该算法融合Katz方法和IMC方法的优点,能够有效缓解遭遇的PU问题的影响,面对极度稀疏的基因-疾病关联数据,能够有效缓解现有方法都会遭遇的数据稀疏性问题。其次,通过引入弹性网正则化缓解数据噪声的影响,在提升预测效果的同时增强算法的容噪性。相较于现有预测方法,KIMC方法预测效果显著提高,同时对于研究者比较关注的新基因和新疾病也能有效预测。该方法对于降低研究成本,帮助研究者深入研究不同疾病的致病基因和基因相关性有很大的意义。
基于本文提出的KIMC方法,后期的研究可以考虑融入更多不同类型的生物数据源,研究如何高效地从这些生物信息中提取关联度更高的基因和疾病的特征信息,以帮助提高预测效果。
猜你喜欢
河北画报(2020年8期)2020-10-27
当代陕西(2019年15期)2019-09-02
学苑创造·A版(2018年11期)2018-02-01
读与写·教育教学版(2017年10期)2017-11-10
雪莲(2017年2期)2017-05-12
环球市场信息导报(2017年1期)2017-04-08
读者(2017年5期)2017-02-15
南都周刊(2015年4期)2015-09-10
南都周刊(2015年3期)2015-09-10
南都周刊(2015年1期)2015-09-10
